设计改造羧酸还原酶合成医药中间体(S)-2-氨基丁醇
2024-02-27张晓辉覃宗敏李聪聪路福平孙周通
张晓辉, 覃宗敏, 李聪聪, 路福平, 曲 戈, 孙周通
(1. 天津科技大学生物工程学院, 天津 300457; 2. 中国科学院天津工业生物技术研究所,天津 300308; 3. 国家合成生物技术创新中心, 天津 300308)
手性胺醇化合物是医药合成中的重要中间体[1-5],目前市场上使用的药物中含有胺醇结构的约占40%[6]。随着手性药物在医药市场上的需求日趋增加,手性医药中间体的绿色生物合成也备受关注。(S)-2-氨基丁醇是一类非常重要的手性砌块,用于众多药物合成中,例如,抗结核杆菌药物乙胺丁醇(Ethambutol)、治疗哮喘的沙美特罗(Salmeterol)[7]、减缓新型冠状病毒并发症的美托洛尔MTP(Metoprolol)[8-9]、治疗增殖性婴儿血管瘤的阿替洛尔(Atenolol)[10-11]、治疗神经源性逼尿肌过度活动NDO的特效药米拉贝隆(Mirabegron)[12]、HIV抑制剂埃替拉韦(Elvitegravur)[2]和一种抗肿瘤药物PDK1抑制剂[13]等。
合成(S)-2-氨基丁醇的化学方法存在一些问题,如需要昂贵的原料和过渡金属催化剂,步骤复杂,区域和对映体选择性不足,并且需要高压设备等。相反,生物催化法因其对环境较为友好、产物的对映选择性高和参与反应时的条件相对温和等优势而备受青睐,是一种很有发展前景的替代性方法。目前生物催化法可利用酰化酶和酰胺酶分解外消旋2-氨基丁醇来获得(S)-2-氨基丁醇[14-15],也可以用转氨酶[16]或氨基酸脱氢酶[17]从2-酮丁酸底物出发,不对称合成(S)-2-氨基丁醇,还有通过胺脱氢酶催化1-羟基-2-丁酮直接胺化合成(S)-2-氨基丁醇[18],这些方法存在转化率低,在合成过程中需要消耗大量氨供体同时产生副产物,底物昂贵等问题。有研究报道,在酵母细胞中构建了一条从苏氨酸出发,通过4步反应合成(S)-2-氨基丁醇的新途径,首次实现(S)-2-氨基丁醇的体内合成。其中,关键一步是利用羧酸还原酶(Carboxylic acid reductase, CAR)催化还原(S)-2-氨基丁酸生成(S)-2-氨基丁醛,但野生型CAR对(S)-2-氨基丁酸的底物特异性较差且催化效率低[19]。
虽然CAR在工业生产醛和各种羧酸的还原衍生物方面具有巨大潜力,受到极大关注,但是该类酶对极性较强的氨基酸类底物活性较差[20]。近年来亦有针对CAR的酶工程研究,但大多聚焦于有机酸类底物[21-22]或者开发基于此类酶的新反应设计[23-24],尚未有针对提升α-氨基酸类底物催化活性的相关文献报道。
研究以Segniliparusrugosus来源的野生型羧酸还原酶SrCAR为研究对象,以N-Boc-(S)-2-氨基丁酸(N-Boc-1a)为模式底物,通过设计提升SrCAR催化效率并级联醇脱氢酶,还原N-Boc-1a为N-Boc-(S)-2-氨基丁醇(N-Boc-1c),最终通过脱Boc保护合成目标产物(S)-2-氨基丁醇(1c),见图1。
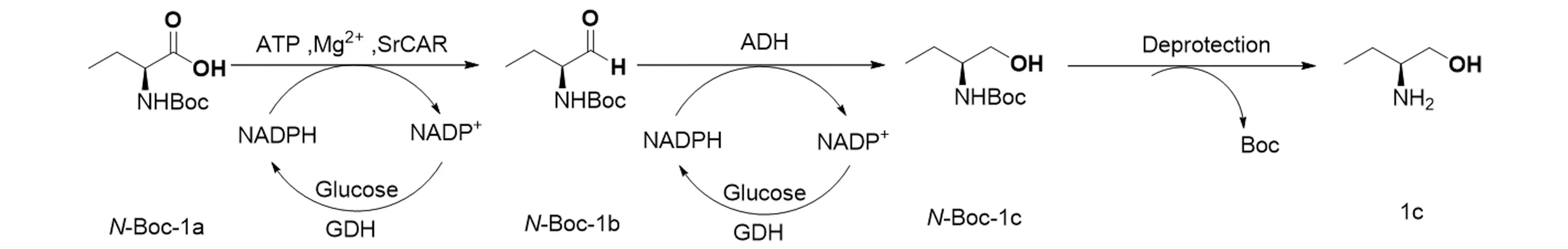
图1 全细胞催化合成 (S)-2-氨基丁醇(1c)Figure 1 Whole-cell synthesis of (S)-2-aminobutanol(1c)
1 材料与方法
1.1 材料
1.1.1 菌种与引物来源
宿主为E.coliBAP1[25];以Segniliparusrugosus来源的羧酸还原酶SrCAR(GenBank:WP_007468889)、荧光假单胞菌(Pseudomonasfluorescens)来源的醇脱氢酶PfADH(GenBank:AF090329.2)和大肠杆菌(Escherichiacoli)来源的醛还原酶Ahr(GenBank:WP_001309160.1)均由武汉金开瑞生物工程公司合成;引物合成均来自安升达(北京)。
1.1.2 试剂与仪器
岛津高效液相色谱仪LC-2030C、岛津高效气相色谱仪LC-2030(日本SHIMADZM公司),其他详见文献[26],N-Boc-(S)-2-氨基丁酸及其他试剂均由喀斯玛平台的生化公司购置。
1.1.3 培养基和培养条件
LB培养基:10 g胰蛋白胨,5 g酵母提取物,10 g氯化钠,1 000 mL纯水;TB培养基:24 g胰蛋白胨,12 g酵母提取物,4 mL甘油,900 mL纯水,100 mL TB缓冲液(2.31 g磷酸二氢钾、16.43 g磷酸氢二钾,100 mL纯水)。将TB培养基与TB缓冲液单独分装灭菌(121 ℃灭菌20 min)备用,使用前将TB液体培养基与TB缓冲液以9∶1的比例混合。
1.2 方法
1.2.1 突变体库的构建
相关突变体及突变体文库均以MegaPCR方法[27]构建,共两轮PCR,以第一轮得到的产物作为第二轮的大引物。PCR扩增体系(50 μL):无菌水(32 μL),10×PCR KOD-Plus-Neo缓冲液(5 μL),模板DNA(1 μL,5 ng),上游引物(1.5 μL,0.3 μmol/L)和下游引物(1.5 μL,0.3 μmol/L),KOD-Plus-Neo DNA聚合酶(1 μL),2 mmol/L dNTPs(5 μL,0.2 mmol/L),25 mmol/L MgSO4(3 μL,1.5 mmol/L)。PCR程序:94 ℃预变性2 min;98 ℃变性15 s,55 ℃退火30 s,68 ℃延伸7 min,以2 kb/min的速率延伸,循环数为30次。PCR产物用限制性内切酶DpnI处理3 h,然后电转到大肠杆菌感受态细胞E.coliBAP1中。如表1所示,根据G430与K524位点组合和K524与A627组合以NNK(反向为MNN)为构建单元设计引物,根据表3阳性位点的氨基酸突变残基,设计G430/K524/E533/A627简并密码子库的引物。

表1 组合突变体文库引物列表Table 1 The primers used in the combinatorial mutant libraries
1.2.2 突变体文库筛选
96深孔培养板中加入300 μL含有卡纳霉素(Kan)(50 μg/mL)的LB培养基。用灭菌后的牙签挑取SrCAR突变体文库的单克隆转移到96深孔培养板中,于37 ℃、800 r/min恒温振荡培养过夜。取120 μL菌液加入到含有60 μL (60%,体积分数)甘油的保菌板中保菌,保菌板用无菌封口膜封上后放置-80 ℃保存。将含有异丙基-β-D-硫代半乳糖苷(IPTG)(0.2 mmol/L)和Kan(50 μg/mL)的700 μL TB补加到96深孔培养板中的剩余菌液中,在30 ℃摇床中诱导培养12 h。收集细胞沉淀并用0.5 mL磷酸盐缓冲液(50 mmol/L,pH 7.4)洗涤菌体两遍,离心后弃上清液。将细胞沉淀在0.5 mL的含有终浓度为5 mmol/LN-Boc-1a、100 mmol/L葡萄糖、2 mg/mL NADPH依赖性葡萄糖脱氢酶(GDH)的粗酶粉、10 mmol/L ATP、10 mmol/L氯化镁(MgCl2)和2 mmol/L NADP+的磷酸盐缓冲液(50 mmol/L,pH 7.4)中重悬菌体,并于30 ℃、800 r/min的条件下反应5 h[28]。设置E.coliBAP1/pET24a为阴性对照,同时设置无菌对照,其他流程均相同。待反应结束后,通过离心收集上清液。取100 μL上清液转移到96孔酶标板(Costar 3603)中,同时加入20 μL溴百里酚蓝[29]。将该96孔酶标板在室温下混匀后,用酶标仪Neo2在615 nm处检测吸光度值,选取吸光度值小于野生型的突变体用于复筛。
以野生型SrCAR为模板,和另外42条已报道的CAR序列[30]进行多序列比对,导出FASTA格式的结果文件,提交到Comulator网站(https://comulator.bio-prodict.com)[31],对G430、E533和K524等3个阳性位点进行共进化分析。
1.2.3 酶活性分析
为进一步筛选出优势突变体,将经过96孔板初筛得到的突变体在250 mL广口锥型摇瓶中扩大培养,进行全细胞生物转化反应的转化率验证。将所得突变体接种至含有Kan(50 μg/mL)的5 mL LB试管中,220 r/min、37 ℃恒温振荡培养10 h。然后,将1 mL菌液转接入含Kan(50 μg/mL)的50 mL TB培养基中,继续培养至菌体OD600值达到0.8左右。随后,以终浓度0.1 mmol/L IPTG以过表达羧酸还原酶,在20 ℃恒温振荡继续诱导培养约15 h。离心后用磷酸盐缓冲液(50 mmol/L,pH 7.4)洗涤菌体两遍,弃上清液,称取湿菌体重量,置于-80 ℃冰箱冷冻保存备用。SrCAR及其突变体生物转化反应的0.5 mL反应体系如下:羧酸还原酶全细胞0.1 g/mL,葡萄糖脱氢酶GDH粗酶粉2 mg/mL,5 mmol/LN-Boc-1a,2 mmol/L NADP+,100 mmol/L葡萄糖,磷酸盐缓冲液(50 mmol/L,pH 7.4),于2 mL EP管中反应,在恒温金属浴中30 ℃,1 000 r/min反应1 h,0.5 h时调节pH 7.4,以12 000 r/min的转速离心3 min收集反应液。取200 μL的上清液于新的2 mL EP管中,加入饱和的NaCl,再加入二倍体积乙酸乙酯400 μL使蛋白质充分变性,并将产物萃取出来,在室温下用恒温振荡混匀仪振荡5 min后,于12 000 r/min、3 min的条件下离心收集上清液。取200 μL上清液过0.22 μm有机膜后进行气相色谱检测[32]。检测条件如表2。
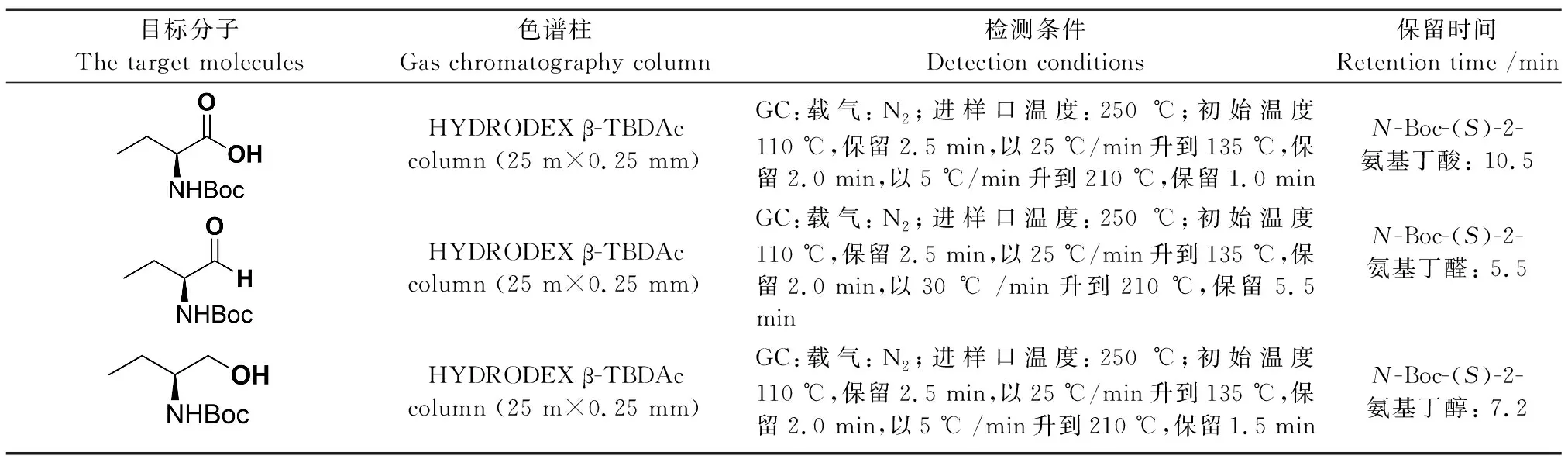
表2 气相色谱检测条件Table 2 Detection conditions of gas chromatography
全细胞催化(S)-2-氨基丁酸的检测:12 000 r/min室温离心5 min收集反应液。取150 μL的上清液于新的2 mL EP管中,加入500 μL乙腈以使其中的蛋白质充分变性,充分振荡后,12 000 r/min离心3 min收集上层反应液。用Marfey’s试剂(14 mmol/L,溶于乙腈,避光备用)衍生[33]:取100 μL乙腈+40 μL碳酸氢钠(1 mol/L)+30 μL衍生剂+50 μL上述上层反应液,在恒温金属浴中于80 ℃、1 000 r/min的条件下衍生10 min。衍生结束后,用10 μL浓盐酸(4 mol/L)淬灭反应。取200 μL反应液用0.22 μm有机膜过滤后进行液相检测。检测条件:液相色谱柱Zorbax SB-C18(4.6 mm×150 mm,5 μm),340 nm处,室温下流速为1 mL/min。进样体积为10 μL,A泵为纯水[含0.1%(体积分数)三氟乙酸],B泵为甲醇[含0.1%(体积分数)三氟乙酸],前3 min以40%的甲醇[含0.1%(体积分数)三氟乙酸]洗脱,4~13 min以60%的甲醇(体积分数0.1%三氟乙酸)洗脱,14~20 min以60%的甲醇(体积分数0.1%三氟乙酸)洗脱。(S)-2-氨基丁酸保留时间为14.3 min,(S)-2-氨基丁醇保留时间为12.5 min。
1.2.4 优势突变体酶学性能表征
在1.2.3节所述0.5 mL反应体系中,30 ℃,1 000 r/min反应,测定在底物浓度0.2~40 mmol/L范围内反应的初速度。经计算得到反应速率和底物浓度,利用GraphPad Prism 9软件进行米氏方程的非线性拟合,得到Km和Vmax,最后计算得kcat[34]。
羧酸还原酶的热稳定性测试:以蛋白质构象受温度影响变性一半时的熔解温度Tm为指标,利用圆二色光谱(Circular dichroism,CD)分析酶蛋白样品的热稳定性[26]。对目的蛋白进行镍柱亲和层析,经脱盐后,用脱盐液将纯酶液稀释到0.2 mg/mL左右,经过高速低温离心的纯酶液上样检测。根据CD仪器记录曲线经拟合后得到Tm。
1.2.5 分子对接与分子动力学模拟
以SrCAR(PDB编号5MSW)[35]为模板,通过在线SWISS-MODEL(https://swissmodel.expasy.org/)同源建模得到XH7的模型。使用Autodock 4.2[36]的拉马克遗传算法(LGA)将底物N-Boc-1a对接到SrCAR与XH7的活性口袋中,将获得的能量最低的复合物结构作为分子动力学模拟的初始结构。研究中模拟了2个体系,分别为野生型SrCAR+N-Boc-1a,突变体SrCAR(XH7)+N-Boc-1a两个复合物结构。所有的分子动力学模拟均使用AMBER 20软件包[37]运行。蛋白质由amber ff14SB力场描述[38],并使用GAFF[39]生成小分子的力场参数。将TIP3P[40]水分子添加到八面体的周期性盒子中进行模拟,距离溶质的距离为1.2 nm,以避免边缘效应的影响。通过加入适当数量的中和离子,使整个系统成为电中性的。用SHAKE算法[41]约束所有涉及氢原子的键,长程静电相互作用采用PME算法[42]来处理,截断半径为1 nm。对2个系统通过最陡下降算法和共轭梯度算法模拟各5 000步,使系统能量最小化。之后,在NVT系综下逐渐加热到298 K,最终,在NPT系综下对每个系统进行了50 ns的模拟,并平行重复3次,所有模拟的时间步长均为2 fs。
1.2.6 产物脱Boc与分离纯化
反应结束后将反应液以4 000 r/min离心30 min,收集上清液,并用磷酸盐缓冲液(50 mmol/L, pH 7.4)洗涤菌体3次,随后在收集到的反应液中加入饱和氯化钠和乙酸乙酯进行萃取,取上层萃取液进行旋转蒸发,再加入二氯甲烷溶解,加入浓HCl(12 mol/L)至pH 2进行脱Boc保护[43],室温反应5 h。反应结束后加入饱和碳酸氢钠淬灭反应,通过分液漏斗分离下层有机溶液,并用无水硫酸钠进行干燥,最后通过旋转蒸发获得(S)-2-氨基丁醇粗提物。用纯水溶解上述固体后,再通过离子交换树脂Doex 50WX8-100对产物进一步提纯[18],获得(S)-2-氨基丁醇纯品。具体操作如下:首先将离子交换柱和产物粗提物分别用5%(体积分数)的硫酸酸化,再用超纯水洗涤柱子,接着以低流速缓慢加入酸化好的样品,用超纯水洗涤至pH 7.0左右,再用100 mL 9%(体积分数)的氨水将样品洗脱,得到的洗脱液经旋转蒸发后得到(S)-2-氨基丁醇。
2 结果与分析
2.1 底物(S)-2-氨基丁酸的N-Boc保护
构建大肠杆菌全细胞催化底物(S)-2-氨基丁酸(1a)反应体系,发现在空载体对照及含有SrCAR重组菌均有87%的底物消耗,且未检测到产物1c[图2(a)]。此外,利用纯酶反应验证,如图2(b)所示,在不含酶的阴性对照反应体系中存在60%的底物消耗,但无产物生成;在含有SrCAR与醛还原酶Ahr纯酶的反应体系中,检测到80%的底物消耗,且有20%的产物(S)-2-氨基丁醇生成。在全细胞催化体系中存在底物1a的背景消耗问题,可能被大肠杆菌内源酶代谢消耗。上述结果表明,SrCAR纯酶能够与大肠杆菌来源的醛还原酶Ahr级联,催化1a生成1c,但是存在明显的底物背景消耗问题,说明1a并非理想底物。
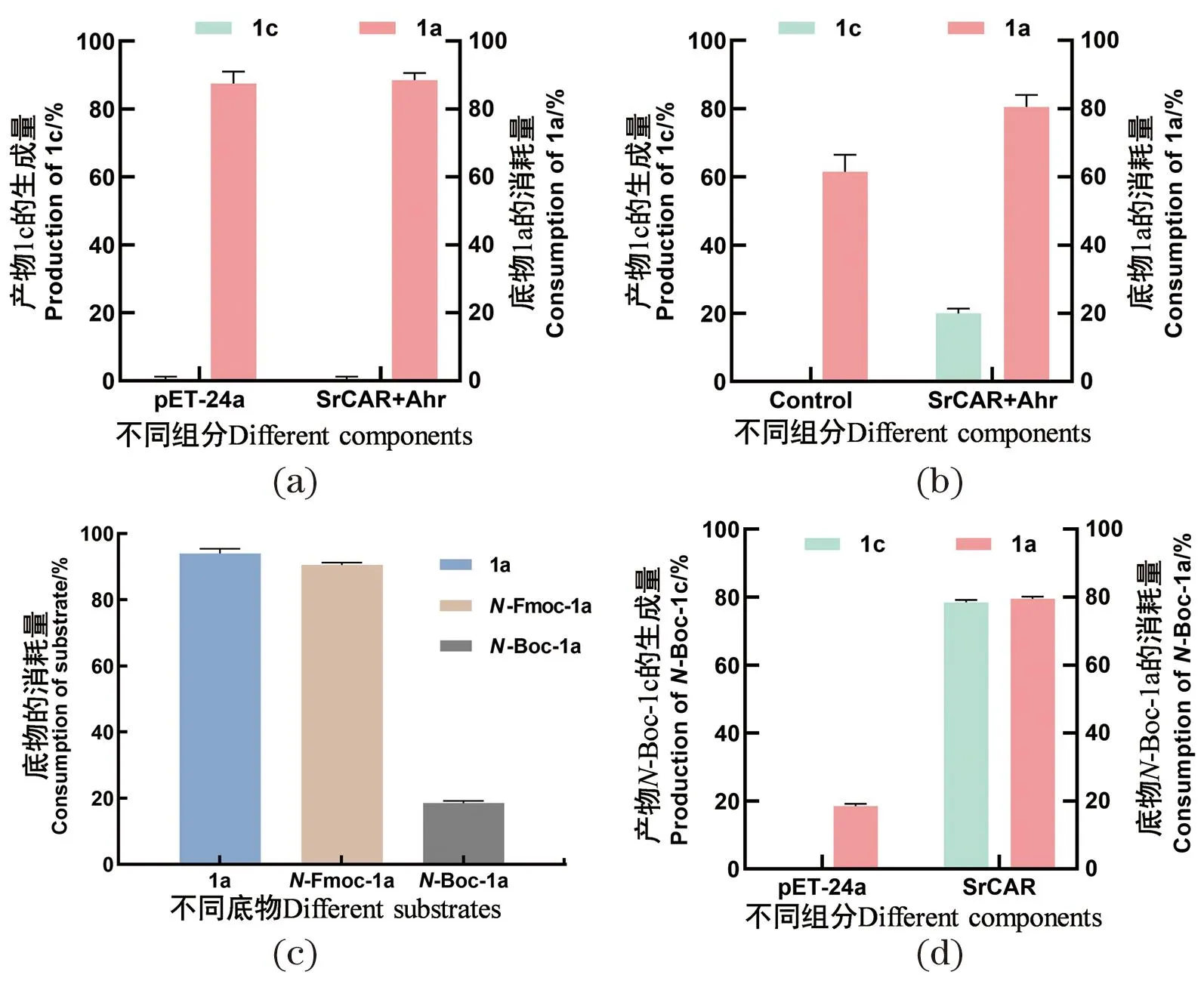
(a)全细胞反应催化底物1a[(S)-2-氨基丁酸],全细胞反应体系为0.1 g/mL的全细胞、NADP+(2 mmol/L)、1a(5 mmol/L)、ATP(10 mmol/L)、Mg2+(10 mmol/L)、GDH(2 mg/mL)和葡萄糖(100 mmol/L),反应条件为1 000 r/min,24 h;(b)纯酶反应催化底物1a[(S)-2-氨基丁酸],纯酶反应体系为SrCAR纯酶(75 μmol/L)、Ahr纯酶(150 μmol/L)、NADPH(10 mmol/L)、1a(5 mmol/L)、ATP(10 mmol/L)和Mg2+(10 mmol/L),反应条件为800 r/min,24 h,Control为不含纯酶但与实验组一致;(c)1a[(S)-2-氨基丁酸]、N-Fmoc-1a和N-Boc-1a在空载全细胞反应体系中的消耗情况;(d)SrCAR对底物N-Boc-1a的底物消耗与产物生成情况;(c)和(d)为全细胞反应底物不同,其他成分同(a),反应时间为1 h。图2 全细胞与纯酶催化还原(S)-2-氨基丁酸反应Figure 2 Reduction of (S)-2-aminobutyric acid by whole cells and purified enzymes
为解决上述背景消耗问题,采用Boc及Fmoc氨基保护1a,即N-Boc-1a和N-Fmoc-1a作为底物,通过全细胞催化体系考察这两种底物的消耗情况。结果如图2(c)所示,在空载(pET24a)体系中,1a的消耗为94%,N-Fmoc-1a的消耗为91%,而N-Boc-1a的消耗仅为18%。说明大肠杆菌内源基因可代谢消耗底物1a及Fmoc保护的底物,但对Boc保护的底物识别能力较差,因此后续研究选择N-Boc-1a作为模式底物。此外,在SrCAR催化体系中,产物生成和底物消耗均为80%[图2(d)],推测在含有SrCAR及其突变体过表达的全细胞反应体系中,可竞争性催化还原模式底物,从而一定程度上避免了内源性的背景消耗。说明对底物1a的-NH2进行Boc保护,能够有效降低背景消耗,并且促进产物生成,因此,后续实验以N-Boc-1a为模式底物开展。
2.2 羧酸还原酶的理性设计
2.2.1 单位点突变体的筛选
以N-Boc-1a为模式底物,基于实验室前期开展的催化机制解析及酶-底物相互作用分析等工作[21,34],对已构建的12个单点饱和突变体文库(T265、S266、G267、H315、S408、G430、T505、D507、Y519、R522、K524、E533)进行全细胞生物转化筛选测试。其中,G430、K524和E533位点上的突变体活性提高幅度最大,突变体G430V、G430K、G430V、K524A、K524Q、E533F等对N-Boc-1a的转化率可达99%(5 mmol/L)。因为这3个位点阳性突变较多且突变体转化率能达到99%,所以理性选取G430、K524和E533为阳性位点。
2.2.2 阳性位点共进化分析
为探索阳性位点G430、E533和K524是否与酶蛋白其他位点存在共同进化关系,对上述3个位点采用在线工具Comulator进行共进化分析。结果表明,只有K524位点存在共进化关联位点A627。因此,对A627位点开展单点饱和突变测试。其中,A627V、A627N和A627L对5 mmol/L底物的转化率可达99%。
2.2.3 组合饱和突变
为进一步提高SrCAR突变体活性,以转化率大于90%为标准,选取G430、K524、E533和A627单点饱和突变体文库中的优势突变残基作为构建单元:G430位点选取的是赖氨酸(K)、色氨酸(W)、异亮氨酸(I)、缬氨酸(V)、谷氨酸(E)以及野生型甘氨酸(G);据此G430位点设计的简并密码子为RWA和KGG。K524位点选择的是丙氨酸(A)、谷氨酰胺(Q)以及野生型赖氨酸(K);对该位点设计的简并密码子为MAA和GCG。E533位点选取的是苯丙氨酸(F)和野生型谷氨酸(E);因没有同时满足需求的简并密码子,所以对该位点设计的密码子为GAA和TTT。A627位点选择的氨基酸为缬氨酸(V)、亮氨酸(L)、天冬酰胺(N)以及野生型丙氨酸(A);设计的简并密码子为KTA、ATT和GCG。以上述简并密码子设计引物构建G430/K524/E533/A627“小而精”的突变体文库(表3),最终从432个转化子中筛选获得优势突变体XH7(G430V/E533F/A627N),转化率达到99%。

表3 阳性位点突变体分析Table 3 Analysis of positive site mutants
为进一步检验获得的系列优势突变体,提升底物N-Boc-1a浓度至20 mmol/L并进行3 h的催化反应。结果如图3所示,突变体A627N、K524F/A627V、G430V/E533F/A627N的生成N-Boc-1c的转化率分别为49%、48%和78%。突变体XH7(G430V/E533F/A627N)的转化率最高,因此后续开展对XH7的动力学参数测定与放大反应分析。

图3 全细胞反应催化还原底物N-Boc-1aFigure 3 Reduction of N-Boc-1a by whole cells
2.3 优势突变体酶学性能表征
为评估SrCAR及其获得的优势突变体XH7的催化效率,对工程化羧酸还原酶的Km、kcat、kcat/Km及比酶活4个参数进行动力学表征。其中,优势突变体XH7的Km值为0.45 mmol/L,与野生型(0.92 mmol/L)相比降低了2倍多,而kcat值与野生型接近,分别为4.41 s-1和4.46 s-1。XH7的催化效率动力学参数kcat/Km为10.21 s-1mmol/L-1,是模板SrCAR(4.78 s-1mmol/L-1)的2.1倍,但比酶活与野生型无明显差异(分别为0.67 U/mg和0.69 U/mg),见表4。因此,优势突变体活性的提高主要在于底物N-Boc-1a与酶的亲和力增加。此外,CD检测结果显示XH7的Tm值比野生型提高了2.3 ℃,说明该突变体在kcat/Km提升的同时,其热稳定性也略有提升(表4)。
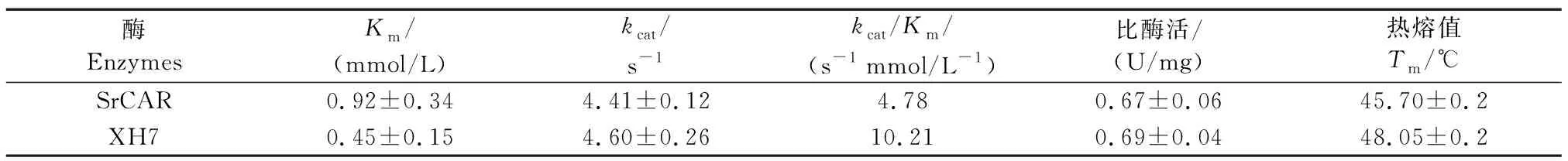
表4 SrCAR优势突变体的动力学和热稳定性参数Table 4 Kinetic parameters and specific activity of SrCAR mutants
2.4 分子对接及分子动力学模拟分析
为阐明突变体催化活性及热稳定性提高的可能分子机制,开展相关计算解析。通过分子对接获得野生型WT及突变体XH7的蛋白-配体-底物复合体结构[图4(a)];接下来对获得的复合体结构开展分子动力学模拟。通过对50 ns的模拟轨迹进行均方根偏差(RMSD)分析发现,XH7的RMSD值分布比野生型更集中,主要富集在0.38 nm附近[图4(b)],说明其在分子动力学模拟过程中结构更稳定。另外,在XH7中627位点由丙氨酸(A)突变为天冬酰胺(N)后,其酰胺侧链可与底物N-Boc-1a形成2个氢键作用(d2和d3),同时也可以与AMP形成氢键相互作用(d4),而在WT中只有骨架N上的氢原子可以与底物形成氢键(d1)[图4(c)]。因此,该位点突变后新形成的相互作用可能更有利于稳定底物N-Boc-1a在口袋中发生活化反应。虽然WT与XH7中430位点的骨架原子与AMP都存在氢键相互作用,但在XH7中甘氨酸(G)突变为缬氨酸(V)后,V430侧链可以与F294形成烷基-π共轭作用(d5),此外,远离活性中心的533位点由谷氨酸(E)突变为苯丙氨酸(F)后,可以与Y604形成π-π共轭效应(d6),由此可知,G430V/E533F突变可能通过与邻近残基的作用而利于提高突变体的热稳定性。进一步分析发生以上相互作用关系的残基间距离随模拟时间变化的趋势可知,G430V/E533F/A627N与底物、辅因子以及关键残基的距离均维持在0.4 nm左右[图4(d)],说明上述相互作用在模拟过程中比较稳定。
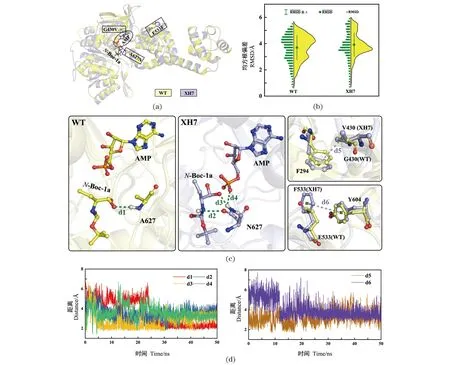
(a)野生型WT及突变体XH7与底物分子N-Boc-1a对接后的复合体结构图;(b)野生型WT及突变体XH7体系的RMSD结果对比;(c)与突变残基G430V/E533F/A627N相关的残基间相互作用分析;(d)相互作用距离在分子动力学模拟过程中随时间变化情况。图4 野生型WT和突变体XH7与N-Boc-1a的分子对接以及复合物的分子动力学模拟结果分析Figure 4 Molecular docking of wild-type WT and mutant XH7 with N-Boc-1a and analysis of molecular dynamics simulation results of the complex
通过在线网站(https://www.expasy.org/resources/protscale)分析突变体XH7与WT的氨基酸疏水性强度。发现突变体XH7与野生型WT相比,430位点由甘氨酸(G)突变为缬氨酸(V)后,以及远离活性中心的533位点由谷氨酸(E)突变为苯丙氨酸(F)后,它们周围的疏水性明显增强,更有利于蛋白结构的稳定,可能也是Tm值略有提高的原因。
2.5 放大反应
为检验工程化的SrCAR能否实现制备级转化,用优势突变体XH7全细胞催化20 mmol/LN-Boc-1a(10 mL反应体系)。检测结果显示中间产物积累较多[图5(a)],推测可能是因为大肠杆菌内源醇脱氢酶(Alcohol dehydrogenase,ADH)还原力不足。据此,利用实验室现有的ADH酶库(共包含90种不同物种来源ADH),以5 mmol/L的中间产物N-Boc-1b为底物筛选合适的ADH,最终选取转化率最高(57%)的荧光假单胞菌(Pseudomonasfluorescens)来源醇脱氢酶PfADH,与突变体XH7建立共表达体系。全细胞催化底物20 mmol/LN-Boc-1a放大反应结果[图5(a)]所示,在PfADH-XH7共表达体系催化反应中,中间产物N-Boc-1b的积累明显减少。在5 h时,底物转化为终产物的量达到99%[图5(b)]。经强酸脱Boc并用离子交换树脂进一步纯化后,对产物进行了NMR分析。结果为1H NMR(400 MHz, CDCl3) δ 3.60(d, J=9.5 Hz, 1H), 3.30(t, J=8.5 Hz, 1H), 2.74(s, 3H), 1.47(dd, J=13.1, 6.4 Hz, 1H), 1.44~1.25(m, 1H), 0.94(t, J=7.1 Hz, 3H)。1H NMR结果分析显示,分离获得的产物为(S)-2-氨基丁醇,其分离得率为60%。
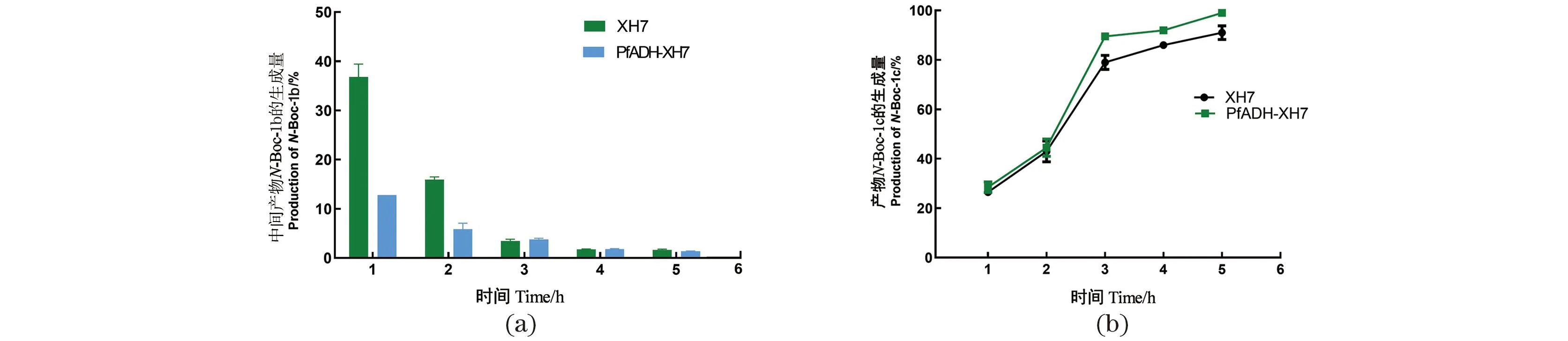
(a)中间产物N-Boc-1b生成情况;(b)产物N-Boc-1c生成情况。图5 XH7与PfADH-XH7中间产物积累与产物生成情况Figure 5 Accumulation and product formation of XH7 and PfADH-XH7 intermediates
3 讨论
(S)-2-氨基丁醇是一类非常重要的手性医药中间体,本研究选择晶体结构已知[35]、催化机制较为清晰[21,34]的SrCAR为模式酶,以N-Boc保护的(S)-2-氨基丁醇(N-Boc-1a)为模式底物。为提高SrCAR催化还原非天然底物N-Boc-1a的活性,通过构建两轮突变体文库并筛选得到优势突变体XH7(G430V/E533F/A627N),其kcat/Km是野生型模板SrCAR的2.1倍。催化活性提升的同时,其热稳定性(Tm值)同步提高2.3 ℃。包含XH7与异源醇脱氢酶PfADH的重组大肠杆菌能够在10 mL反应体系中5 h内完成底物N-Boc-1a(20 mmol/L)的转化,转化率达到99%,并经一步脱保护,获得终产物(S)-2-氨基丁醇。反应体系中所用的ATP比较昂贵,后期可采用磷酸激酶实现由AMP到ATP再生,从而进一步提高放大体系的应用潜力。综上,研究为生物合成(S)-2-氨基丁醇提供新思路,也为羧酸还原酶催化氨基酸类底物提供了理论参考。
