SOCS3在病理性疼痛中的作用
2024-02-27易小苏王冬梅
易小苏, 王冬梅
(福建师范大学生命科学学院,福建省发育与神经生物学重点实验室, 福州 350117)
急性疼痛属于身体的报警系统,机体可在组织损伤的即刻作出反应,避免进一步伤害。慢性疼痛是指反复发作持续超过1个月的疼痛,病理性疼痛属于慢性疼痛,发生于影响外周或中枢神经系统内感觉通路的病变或疾病[1, 2]。病理性疼痛由于其持续存在,严重影响患者生活质量,病理性疼痛的发生机制尚未弄清,因此,目前临床仍无恰当的治疗方法。近年的研究发现,细胞因子信号传导抑制因子3(suppressor of cytokine signaling 3,SOCS3)在病理性疼痛及吗啡耐受中发生表达的变化,通过负性调控多种细胞因子及信号通路发挥镇痛作用。
1 细胞因子信号传导抑制因子3的结构与功能
SOCS3是细胞因子信号传导抑制因子蛋白质家族(suppressor of cytokine signaling protein family,SOCS)的一员[1, 3]。SOCS蛋白质家族包括8个成员(SOCS1~SOCS7和CIS)[2, 3]。SOCS3在N-末端有一个激酶抑制区(KIR),中间有一个Src同源2区(SH2),C末端区有一个高度保守的域称为SOCS盒[4, 5]。SOCS盒大约由40个氨基酸残基组成,包含2个保守区,分别称为BC盒和Cul5盒[5]。细胞在受到一些生长因子和细胞因子刺激时,SOCS3的酪氨酸被磷酸化,并且只有204位和221位的酪氨酸被磷酸化,而这2个位点都定位在保守的SOCS盒[6]。SOCS3可以被泛素-蛋白酶体系统快速降解[7]。SOCS盒的N末端部分(1~12残基)与elonginBC 结合,SOCS盒的C末端部分与滞蛋白(cullin)结合(滞蛋白形成E3泛素连接酶支架),SOCS盒通过与elonginBC和cullin蛋白结合,促进E3泛素连接酶的形成,诱导与SOCS3结合的蛋白质的降解[8]。SOCS3在多个水平上负调控CCAAT/增强子结合蛋白质β(CCAAT/enhancer-binding proteinβ,C/EBPβ),降低C/EBPβ蛋白质和mRNA表达,加速C/EBPβ蛋白质降解[9]。
细胞因子与细胞膜上受体结合,导致细胞发生一系列活动,而细胞因子介导的信号及时消失,避免信号过度激活,对细胞存活也是必要的,SOCS家族就是这样一类抑制细胞因子介导信号传导的蛋白质家族[10]。在未受刺激的细胞中大多数SOCS基因的表达水平非常低或无法检测,外界条件例如肿瘤坏死因子(tumour necrosis factor,TNF)或脂多糖(lipopolysaccharide,LPS)刺激下会诱导SOCS表达,过度表达的SOCS再抑制细胞因子介导的信号传导[11]。在γ干扰素(interferon γ,IFNγ)、LPS刺激或泰勒鼠脑脊髓炎病毒(Theiler’s murine encephalomyelitis virus,TMEV)感染的小胶质细胞中,加入维生素D3的活性分子1,25(OH)2D3或25(OH)D3,以IL-10依赖方式增加SOCS3表达,从而降低感染导致的促炎因子例如IL-6、IL-12及诱导型一氧化氮合酶(inducible nitric oxide synthase,iNOS)的表达[12]。
SOCS3参与机体各种关键过程,包括免疫、生长、造血和新陈代谢[9],SOCS3功能失调导致多种疾病,例如免疫系统疾病、血管炎性疾病、肥胖与胰岛素抵抗和癌症等[10]。在合成髓磷脂少突胶质细胞糖蛋白肽35-55(syntheticmyelin oligodendrocyte glycoprotein peptide 35-55,MOG35-55)诱导的实验性自身免疫性脑脊髓炎(experimental autoimmune encephalomyelitis,EAE)小鼠模型中,雷帕霉素治疗会导致EAE小鼠SOCS3表达增加,提示雷帕霉素可能通过增加SOCS3表达发挥免疫抑制作用[13]。用腺病毒载体感染神经干细胞过表达SOCS3,抑制胶质纤维酸性蛋白质(glial fibrillary acidic protein,GFAP)生成,说明SOCS3会抑制神经干细胞向星形胶质细胞分化[14]。SOCS3在胆道系统恶性肿瘤(biliary tract cancer,BTC)组织中的阳性表达明显低于癌旁组织,进一步分析发现,SOCS3的表达与BTC肿瘤分化程度和TNM分期有关,提示SOCS3的低表达可能参与了BTC的增殖分化、侵袭和转移等恶变过程,SOCS3可能为BTC的抑癌基因[15]。而在糖尿病患者的血浆中SOCS3的表达明显比空腹血糖受损(impaired fasting glucose,IFG)/葡萄糖耐量降低(impaired glucose tolerance,IGT)患者,或正常的葡萄糖耐量(normal glucose tolerance,NGT)人群高[16]。
2 细胞因子信号传导抑制因子3在神经病理性疼痛的作用
SOCS3能通过调控下游因子和负反馈上游因子,在神经病理性疼痛中发挥镇痛作用。并且SOCS3能在RNA层面被调控,在神经病理性疼痛中发挥镇痛作用。
2.1 SOCS3负反馈抑制JAK/STAT3信号通路缓解神经病理性痛
细胞用白血病抑制因子(leukemia inhibitory factor,LIF)刺激,LIF与糖蛋白130(glucoprotein 130, gp130):LIFR异二聚体结合,诱导相连的JAK1的自磷酸化,磷酸化的JAK1(phosphorylatedp Janus kinase 1,pJAK1)再磷酸化受体的酪氨酸,STAT3结合到gp130上磷酸化的酪氨酸,被磷酸化,使其二聚化并转移到细胞核内,诱导SOCS3的转录,通过SH2区结合到gp130上757位磷酸化酪氨酸,SOCS3与pJAK1结合,复合物从受体复合物上释放,因此,STAT3不能被pJAK1磷酸化,接着SOCS3泛素化pJAK1,pJAK1以SOCS3的SOCS盒依赖方式被蛋白酶体降解[5, 10, 11]。SOCS3负反馈抑制JAK/STAT3信号通路机制如Fig.1所示。SOCS3可以通过两个相邻的结合表面同时与JAK2和受体相互作用,形成高亲和力的三元复合物,增加SOCS3对JAK和细胞因子的抑制作用,而缺失受体的情况下削弱SOCS3对JAK的抑制作用[17]。SOCS3抑制JAK对底物磷酸化的原因为JAK插入环最后3个残基GQM,突变GQM这一序列完全消除了SOCS3对JAK2的抑制能力,SOCS3与JAK2的结合可能改变GQM与JAK2底物结合位点的相对位置,从而影响JAK2对底物的磷酸化[17]。SOCS3是JAK2的非竞争性抑制剂(JAK的底物为ATP和含酪氨酸的底物),这一非竞争性抑制作用与ATP竞争性抑制剂相比有明显优势,因为体内ATP浓度很高,并且酪氨酸激酶ATP结合位点结构相似[17]。
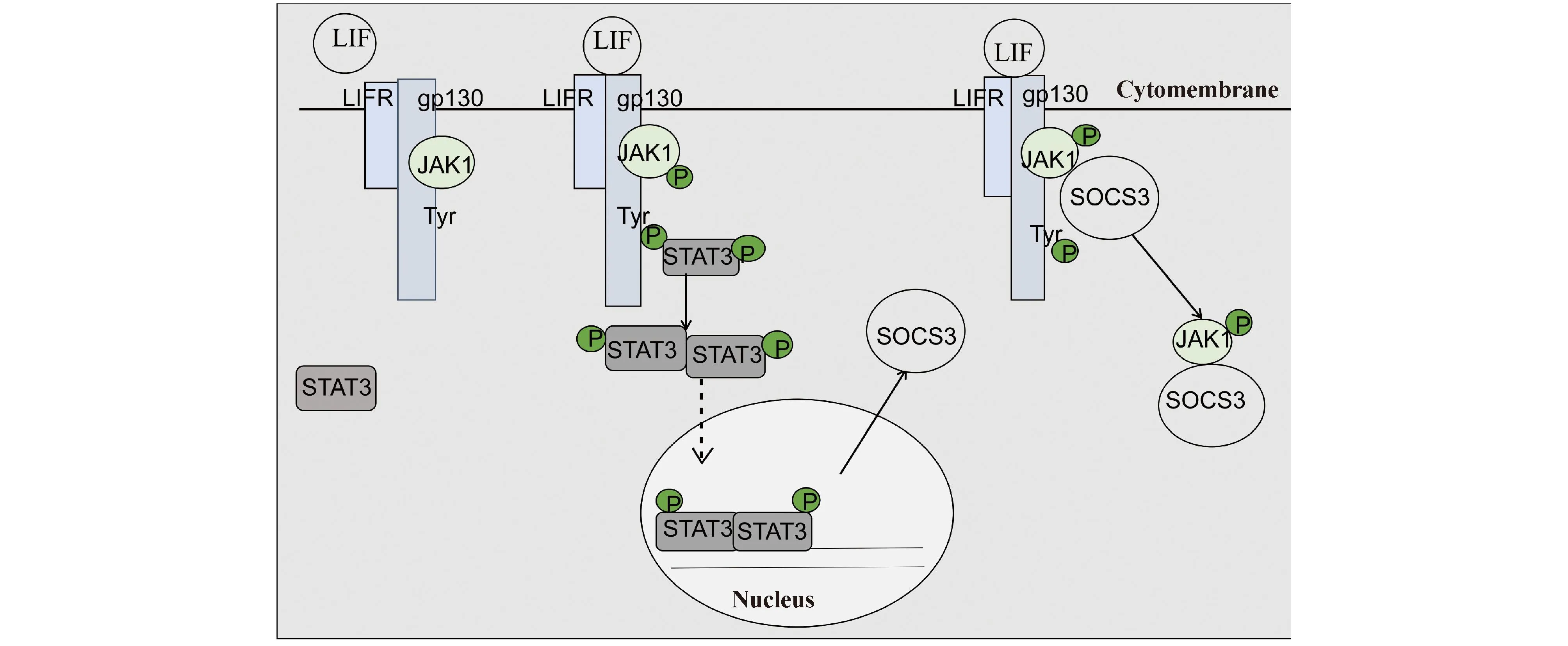
Fig.1 SOCS3 inhibits the JAK/STAT3 signaling pathway in a negative feedback way[5] LIF binds to the receptor, which induces the auto-phosphorylation of JAK1 that is attached to the receptor, then pJAK1 phosphorylates the tyrosine of the receptor. STAT3 binds to the phosphorylated tyrosine of the receptor. It is then phosphorylated, dimerized and later translocates into the nucleus and induces the expression of SOCS3. SOCS3 binds to the phosphorylated tyrosine of the receptor, then binds to pJAK1 and dissociates from the receptor, so STAT3 cannot be phosphorylated by pJAK1
在慢性压迫损伤(chronic constriction injury,CCI)模型中同侧脊髓背角SOCS3表达增加,可能是磷酸二聚化的STAT3转移到细胞核内诱导SOCS3转录,而SOCS3的表达会抑制磷酸化的STAT3水平,发挥镇痛作用。例如在CCI模型大鼠中脊髓背角Janus激酶/信号转导子和转录激活子3(Janus kinase/signal transducer and activator of transcription 3,JAK/STAT3)信号通路激活,鞘内注射慢病毒载体介导SOCS3过表达,其显著降低脊髓背角中磷酸化的STAT3水平,抑制CCI诱导的IL-6、CC趋化因子配体2(CC chemokine ligand 2,CCL2)和激活转录因子3(activating transcription factor 3,ATF3)的表达,进而缓解CCI诱发的机械接触诱发疼痛[18]。鞘内注射阿司匹林触发的脂蛋白A4(aspirin-triggered lipoxin A4,ATL),为脂蛋白A4(lipoxinA4,LXA4)的差向异构体,促进CCI大鼠脊髓中SOCS3 mRNA进一步表达,抑制CCI大鼠脊髓中磷酸化的STAT3(phosphorylatedp signal transducer and activator of transcription 3,pSTAT3)的增加,抑制CCI诱导的炎性因子IL-1β、IL-6和肿瘤坏死因子α(tumour necrosis factor-α,TNF-α)的表达,从而缓解CCI机械接触诱发的疼痛[19]。双侧坐骨神经慢性压迫损伤(bilateral chronic constriction injury,bCCI)大鼠鞘内注射pcDNA-阴阳1(yin yang 1,YY1)表达载体,使YY1(阴阳1)过表达,其增加脊髓中SOCS3的mRNA和蛋白质的表达,降低pSTAT3的表达,降低炎症因子 IL-1β和IL-6的分泌,缓解CCI大鼠的机械接触诱发的疼痛、热痛觉过敏和冷痛觉过敏[20]。鞘内注射WP1066(STAT3抑制剂)显著抑制bCCI大鼠脊髓中上调的SOCS3的mRNA和蛋白质、抑制JAK2的mRNA和蛋白质表达,下调STAT3的mRNA和磷酸化水平,缓解bCCI大鼠的机械接触诱发的疼痛、热和冷痛觉过敏[21]。
2.2 非编码RNA调节SOCS3表达缓解神经病理性痛
长非编码RNA(long noncoding RNA,lncRNA)能调节SOCS3表达,在疼痛模型中负调控某些lncRNA,会增加SOCS3表达发挥镇痛作用。例如在 bCCI大鼠脊髓中长非编码RNA DILC(long noncoding RNA DILC,lncRNA DILC)表达增加,通过鞘内注射lncRNA DILC siRNA,降低 lncRNA DILC表达,会导致脊髓中SOCS3的表达增加,抑制STAT3的磷酸化,减轻bCCI大鼠的机械接触诱发的疼痛、热痛觉过敏和冷触诱发的疼痛[1]。体外小胶质细胞分别转染pcDNA-DILC和DILC siRNA,也证实lncRNA DILC能通过调节SOCS3的表达,调节JAK/STAT3信号通路[1]。bCCI组大鼠鞘内注射lncRNA-linc00311和lncRNA-AK141205的siRNA能抑制pSTAT3的表达,缓解bCCI大鼠的机械接触诱发的疼痛和热与冷痛觉过敏;细胞用WP1066(STAT3抑制剂)治疗后lncRNA-linc00311和lncRNA-AK141205表达下调[22],表明lncRNA-linc00311和lncRNA-AK141205可能在STAT3的上游发挥致痛作用。
微RNA(microRNA,miRNA)为基因表达的关键转录后调节因子,microRNA-30a-5p能在转录水平负调控SOCS3[23]。在疼痛模型中抑制某些miRNA表达,会增加SOCS3表达发挥镇痛作用。例如miR-218作用于SOCS3的3′非翻译区并下调SOCS3表达,在CCI模型大鼠的脊髓中miR-218表达上调,鞘内注射miR-218抑制剂,上调CCI大鼠脊髓中SOCS3 mRNA和蛋白质表达,显著降低CCI大鼠脊髓中STAT3的磷酸化,抑制CCI诱导的炎症因子(IL-1β、IL-6和TNF-α)的表达,缓解CCI诱发的机械接触诱发痛和热痛觉过敏[24]。在糖尿病周围神经病变(diabetic peripheral neuropathy ,DPN)模型大鼠中通过抑制miR-221表达,降低miR-221对SOCS3表达的负调控作用,增加SOCS3表达发挥镇痛作用[25]。这些现象提示,非编码RNA通过上游调控SOCS3作用参与神经病理性痛的发生与发展。
3 细胞因子信号传导抑制因子3在其它病理性疼痛的作用
SOCS3不仅在神经病理性疼痛中发挥作用,还在炎性疼痛、骨癌痛、足底切口痛和吗啡耐受中发挥镇痛作用。
3.1 SOCS3通过抑制多种炎性因子介导的信号通路缓解炎性痛
在炎性疼痛模型中增加SOCS3表达能发挥镇痛作用。例如完全弗氏佐剂(complete Freund’s adjuvant,CFA)诱导的炎性疼痛大鼠,PVN内SOCS3在急性期蛋白质表达水平增加,慢性期表达下降,PVN内注射SOCS3-腺相关病毒(adeno-associated virus,AAV),增加慢性期SOCS3表达量并降低PVN内c-fos(神经元兴奋标志)阳性细胞数量,减轻CFA诱导的慢性期机械接触诱发的疼痛和热痛觉过敏[7]。在胶原诱导关节炎模型的大鼠,鼠耳草乙酸乙酯提取物通过增加SOCS3表达,抑制JAK/STAT信号通路发挥抗炎镇痛作用[26]。SOCS3能抑制多种炎性因子介导的信号通路,这可能是SOCS3缓解炎性痛的原因。例如在BV-2细胞中用ATP敏感性钾通道(ATP-sensitive potassium channel,KATP)打开剂克罗卡林预处理,增加SOCS3表达并且以SOCS3依赖方式,抑制LPS、IL-1β和IL-6介导的炎症反应,发挥抗炎作用;并且克罗卡林以SOCS3依赖方式缓解IL-1β介导的小鼠机械性痛觉超敏[27]。在体外研究中,SOCS3通过抑制TNF受体相关因子6(TNF receptor-associated factor 6,TRAF6)的磷酸化,抑制TGFβ活化激酶1(TGFβ activated kinase1,TAK1)的激活,进而负调控IL-1介导的信号通路[28]。在成骨细胞中,LPS瞬时诱导SOCS3表达,抑制LPS诱导的IL-6的分泌,SOCS3的KIR、SOCS盒的204位酪氨酸和SH2区对SOCS3抑制IL-6的分泌是必不可少的;SOCS3通过抑制转录因子C/EBPβ而不是核因子κB(nuclear factor kappa-B,NF-κB)结合到IL-6启动子区,继而抑制IL-6表达[9]。miR-218抑制剂能抑制IL-6刺激的体外培养的初级小胶质细胞中STAT3的磷酸化,抑制STAT3信号通路下游基因环氧合酶2(cyclooxygenase 2,COX2)和CCL2的表达[24]。SOCS3基因条件性敲除小鼠的肝和巨噬细胞在用IL-6刺激后,可延长pSTAT3表达的时间[29]。在LPS刺激的心肌细胞中,苯丙硝唑以SOCS3依赖方式抑制kappa B 抑制因子激酶(inhibitor of kappa B kinase,IKK)磷酸化和IκBα(NF-κB抑制剂)降解,从而抑制NF-κB的激活和核转移[30]。在BV-2细胞中过表达SOCS3能减少LPS诱导的IL-6、 IL-1β和TNF-α的表达,能降低LPS诱导的p65和p38的磷酸化[2]。这些现象提示,SOCS3缓解炎性疼痛的机制涉及多种细胞因子、趋化因子及转录因子。
3.2 SOCS3在骨癌痛、足底切口痛和吗啡耐受中的作用
在骨癌痛、足底切口痛、吗啡耐受模型中增加SOCS3表达发挥镇痛作用。例如在骨癌痛模型大鼠同侧腰2~5背根神经节中SOCS3蛋白质水平显著下降,免疫组化显示,SOCS3在背根神经节(dorsal root ganglia,DRG)中主要定位于神经元,鞘内注射慢病毒载体SOCS3增加背根神经节中SOCS3蛋白质水平的表达,抑制背根神经节神经元的兴奋性和背根神经节内TLR4的过表达,缓解肿瘤细胞注射同侧后肢机械痛觉过敏[4]。在足底切口痛模型小鼠中,鞘内给药KATP打开剂克罗卡林,以Gas6/Axl信号通路依赖方式增加脊髓中SOCS3表达发挥镇痛作用[27]。足底切口模型小鼠腹腔或鞘内注射芍药苷,能通过增加脊髓中热休克蛋白70(heat shock protein 70,HSP70)表达,作用于细胞上的TLR4,增加脊髓中SOCS3表达,发挥抗炎镇痛作用[31]。吗啡是治疗疼痛的有效药物,但长期使用吗啡治疗疼痛会出现吗啡耐受的现象,吗啡的镇痛效力下降或需加大吗啡用量。在吗啡耐受小鼠中,利多卡因通过增强小鼠脊髓中SOCS3表达缓解小鼠吗啡耐受,并且在BV-2细胞中证实,利多卡因以SOCS3依赖方式抑制吗啡诱导的IL-1β和TNF-α的mRNA增加,抑制吗啡诱导的NF-κB的核转移,并且在BV-2细胞中利多卡因以钙依赖性蛋白激酶β(calcium-dependent protein kinase kinase β,CaMKKβ)-腺苷5′-单磷酸激活蛋白激酶(adenosine 5′-monophosphate (AMP)-activated protein kinase,AMPK)-依赖方式上调细胞中SOCS3的表达[32]。microRNA-30a-5p能在转录水平负调控SOCS3表达,AMPK激活剂二甲双胍通过抑制microRNA-30a-5p表达增加SOCS3表达缓解小鼠吗啡耐受[23]。
4 问题与展望
本文综述了SOCS3的结构、功能、在疼痛中的作用及机制。SOCS3通过负反馈抑制JAK/STAT3信号通路及下游基因的表达,阻碍 IL-1、IL-6和TNF-α等多种炎性因子的分泌,调控NF-κB的激活和核转移等,发挥抗炎镇痛作用。疼痛产生的机制错综复杂,包括多种细胞因子之间的相互作用,不同的镇痛药物通过不同机制对不同或相同的细胞因子产生作用发挥镇痛作用。通过特异microRNA精准调控SOCS3表达,会进一步调控特异性细胞因子或作为SOCS3治疗疼痛的一个潜力研究方向。
