NLRP3炎症小体在阿尔兹海默症中的作用及潜在治疗靶点
2024-02-27秦合伟李彦杰
高 洋, 秦合伟,2)*, 李彦杰,2)
(1)河南中医药大学 康复医学院, 郑州 450046;2)河南中医药大学第二附属医院 康复医学科, 郑州 450000)
阿尔茨海默症(Alzheimer’s disease, AD)是痴呆的主要原因,发生于老年和老年前期,以进行性认知障碍为特征的中枢神经退行性疾病,其主要临床表现为顺行性情景记忆障碍、视空间、语言能力损害、执行功能障碍及人格行为改变[1]。研究表明,AD的发病因素与环境和遗传有关,但最关键的危险因素是年龄,随着全球社会老龄化问题的日益突出,阿尔茨海默病发病率呈逐年增加趋势,为家庭、个人带来巨大经济负担[2]。大脑中β-淀粉样蛋白沉积(β-amyloid, Aβ)、微管相关蛋白tau磷酸化(tauphosphorylation)引起的神经纤维缠结及神经元丢失是AD最主要病理特征[3]。目前,神经炎症被认为在AD的发病机制中发挥核心作用[4],NLRP3(nucleotide-binding oligomerization domain-like receptor pyrin domain-containing 3, NLRP3)炎症小体感知Aβ、tau蛋白聚集触发炎症信号介导慢性炎症及细胞焦亡,促进AD生理病理变化,从而导致认知能力下降[5],揭示 NLRP3炎症小体的激活在诱导AD炎症反应及病理特征发挥关键作用,此外,有研究证实,针对NLRP3炎症小体的抑制性治疗减轻认知障碍具有较大潜力[6]。因此,本文对NLRP3炎症小体影响AD的发病机制,以及靶向治疗AD的临床策略进行综述,以期为临床治疗AD提供新思路。
1 NLRP3炎症小体的结构及活化
NLRP3炎症小体是目前研究最多的炎症小体,亦与神经系统疾病关系密切,其作为感知有害刺激的传感器启动固有免疫应答从而扩大炎症级联反应,然而NLRP3炎症小体的结构及激活途径复杂,且确切激活机制尚不明确。
1.1 NLRP3炎症小体的结构
NLRP3炎症小体是一种多聚体细胞溶质蛋白质复合物,是先天免疫系统的重要组成部分,其通过模式识别受体(pattern recognition receptors,PRR)家族中NLRs感知病原体相关分子模式(pathogen associated molecular patterns, PAMPs)和损伤相关分子模式(damage-associated molecular patterns, DAMPs)进行组装和激活,从而引发促炎因子释放。NLRP3炎症小体由细胞质传感器分子NLRP3,衔接蛋白细胞凋亡相关斑点样蛋白(apoptosis speck-like protein containing a caspase recruitment domain, ASC)和胱天蛋白酶1前体(pro-caspase-1)组成,其中NLRP3发挥主导作用。NLRP3是具有模式识别功能的NOD(nucleotide oligomerization domain,NOD)样受体蛋白质,且氨基端含有pyrin结构域(pyrin domains,PYD),ASC是炎症小体内聚蛋白质,在NLRPs和胱天蛋白酶1前体之间架起相互作用的桥梁,其氨基端通过PYD-PYD相互作用招募NLRP3,ASC羧基端含有半胱天冬氨酸蛋白酶募集结构域(caspase activation and recruitment domain,CARD)并与胱天蛋白酶1前体中的CARD结构域相匹配[7],炎症小体寡聚形成胱天蛋白酶1激活平台,活化的胱天蛋白酶1介导白介素-1(interleukin-1β, IL-1β)和白介素-18(interleukin-18,IL-18)促炎因子释放,从而诱导炎症反应发生[8]。活化的胱天蛋白酶1还可以裂解gasdermin D,诱导非凋亡形式的裂解性程序性坏死(Fig.1)。
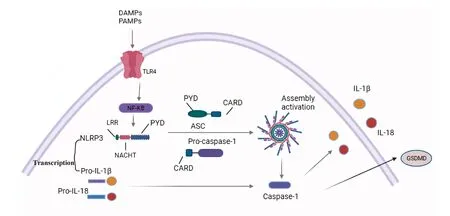
Fig.1 Structure of NLRP3 inflammasome Activation of NLRP3 inflammasome involves the assembly of NLRP3 inflammasome components (NLRP3, ASC, Pro-caspase-1) to form a complete NLRP3 inflammasome complex; LRR-Leucine-rich repetitive structural protein;NACHT-Central nucleotide binding and oligomerization domains with ATPase activity;PYD-The N-terminal Pyrin domain
1.2 NLRP3炎症小体的活化
目前认为,炎症小体的激活有两种信号通路,一种是包括启动和激活两步骤的经典活化途径,一种是独立于Toll 样受体 4(Toll-like receptors 4,TLR4)的非经典信号通路。后者感染革兰氏阴性菌的胞质脂多糖是非典型炎症小体激活的常见分子,人胱天蛋白酶4、胱天蛋白酶5 和鼠胱天蛋白酶11感知脂多糖后互相结合触发其激活和寡聚,进而诱导NLRP3炎症小体活化[9]。经典活化途径中巨噬细胞静息状态下NLRP3、IL-1β的浓度不足以激活炎症小体,而启动步骤中,TLRs识别DAMPs和PAMPs或细胞因子受体(例如肿瘤坏死因子TNF受体)刺激核转录因子κB(nuclear factor κB, NF-κB)传导启动信号,并诱导NLRP3和pro-IL-1β转录增加其表达[10, 11]。激活阶段由晶体和颗粒物质、细菌、ATP等诱导炎症小体组装和活化,而离子稳态、溶酶体裂解、线粒体活性氧(mitochondrial oxygen species, mtROS)生成及功能障碍等影响因素也参与调控NLRP3炎症小体激活。因此,本文讨论上述影响因素调控NLRP3活化的作用机制。
1.2.1 离子稳态 细胞内K+外流是诱导NLRP3活化的重要信号途径。胞内K+浓度降低是激活NLRP3炎症小体的特定上游,细菌毒素和颗粒物强烈触发K+外排并伴随NLRP3依赖性的IL-1β分泌[12]。NIMA(Never-in-mitosis A,NIMA)相关蛋白激酶7(NIMA-related kinase 7, Nek7)作为NLRP3的结合蛋白质,也依赖于K+外排促进NLRP3-Nek7相互作用调节NLRP3的寡聚和激活[13]。进一步研究机制表明,神经胶质细胞大量表达的嘌呤能受体(purinergic receptor, P2X7)作为ATP激活的阳离子通道,激活后改变细胞膜通透性介导K+外流从而活化NLRP3炎症小体[14]。穿过质膜的K+外排是炎症小体激活基本机制,而K+外排通道也至关重要。巨噬细胞中的TWIK2(two-pore domain weak inwardly rectifying K+channel 2)与P2X7受体(P2X7purinergic receptor, P2X7R)协同调节NLRP3激活,后者使Ca2+内流改变膜电位,驱动K+通过前者外流[15]。此外,双孔钾通道 THIK-1(the K channel tandem pore domain halothane-inhibited potassium channel 1,THIK-1)作为特异性调节因子,同样调节P2X7受体介导的ATP依赖性NLRP3激活和释放IL-1β[16]。因此,K+外流是激活NLRP3炎症小体的重要因素。另有研究证明,K+外排是驱动Ca2+内流的关键,且二者在NLRP3激活中协同作用。ATP诱导膜孔pannexin-1募集激活P2X7受体引发K+外流,继而引发Ca2+内流导致线粒体膜电位失衡和mtROS生成,从而激活NLRP3炎症小体[17]。据报道,钙动员也是NLRP3炎症小体激活的关键因素[18],但其在NLRP3炎症小体活化中的作用有待进一步研究。信号依赖的Ca2+内流和内质网Ca2+释放导致胞质内Ca2+水平快速增加,线粒体钙单转运体(mitochondrial calcium uniporter,MCU)摄取过量Ca2+导致线粒体代谢紊乱,mtROS生成增加,并且MCU通过抑制吞噬溶酶体膜修复,二者共同促进炎症小体激活[19]。然而,有研究表明,BAPTA作为一种强Ca2+螯合剂,可以独立于其作为螯合剂的功能抑制NLRP3炎症小体的活化[20]。综上可知,NLRP3炎症小体激活涉及K+外流和Ca2+内流,而胞质钾降低是直接针对质膜通透性诱导炎症小体组装的重要性信号,Ca2+流动在炎症小体活化中的作用存在争议。
1.2.2 溶酶体裂解 二氧化硅和明矾激活NLRP3炎症小体需要吞噬作用,晶体吞噬导致溶酶体破裂和组织蛋白酶B的释放,从而导致胱天蛋白酶1活化以及随后IL-1β的裂解成熟[21]。有研究显示,低水平的溶酶体不稳定激活NLRP3与质膜阳离子通量也相关,溶酶体膜透化期间释放的溶酶体衍生因子介导K+外流、Ca2+内流引发NLRP3/ASC炎症小体组装活化[22]。此外,颗粒物质诱导组织蛋白酶的释放不仅有助于IL-1β的裂解成熟,而且促进IL-1β前体的合成,揭示溶酶体失稳参与炎症小体活化的启动和激活步骤[23]。最近研究证明,小胶质细胞表达的P2X7受体,高水平ATP刺激可增加溶酶体PH值和减缓自噬体和溶酶体融合,从而降低细胞外碎片清除能力,并介导溶酶体组织蛋白酶B激活NLRP3炎症小体[24]。尽管以上研究证明,溶酶体裂解可激活NLRP3炎症小体,但仍需进一步探究具体机制。
1.2.3 线粒体功能障碍及自噬 线粒体是动态双膜结合细胞器,除为细胞生物合成提供所需的底物和能量外还调节细胞凋亡和传导钙信号等,大量研究证明,线粒体在NLRP3炎症小体激活中至关重要。NLRP3激活剂致线粒体功能紊乱不能直接介导炎症小体激活,而是释放mtROS、线粒体DNA(mitochondrial DNA, mtDNA)等衍生因子作为DAMPs激活NLRP3。mtROS的释放与炎症小体活化密切相关。Han等人[25]研究发现,线粒体呼吸链紊乱生成mtROS,硫氧还蛋白相互作用蛋白(thioredoxin-interacting protein,TXNIP)经ROS氧化后从硫氧还原蛋白(thioredoxin,TXr)中脱离,TXNIP与NLRP3结合诱导炎症小体活化、促进IL-1β分泌,而靶向ROS的抗氧化剂MitoQ可以逆转这一过程,减少炎症损伤。此外,静息NLRP3定位于内质网和细胞质中,当病毒和细菌感染以及非感染性刺激激活存在时,NLRP3和ASC转移到线粒体相关膜中,同时后者介导Ca2+从内质网向线粒体转移,过量Ca2+诱导mtROS生成刺激NLRP3活化[26]并驱动炎症级联反应,促进小胶质细胞释放促炎因子并正反馈ROS的生成。mtROS的过度生成会导致线粒体完整性破坏,线粒体内容物mtDNA通过线粒体外膜透化、线粒体通透性过渡孔等途径释放到细胞质中激活NLRP3炎症小体,但具体机制仍待研究[27]。
线粒体自噬是一种选择性自噬机制,通过去除受损线粒体减少线粒体应激对生物体的损害[28]。上述表明线粒体功能障碍诱导的一系列应激反应,包括膜电位改变、线粒体动力学改变、mtDNA释放等作为NLRP3炎症小体的激活信号,而增加线粒体自噬是调节NLRP3活化的重要调节因子[29]。丝氨酸-苏氨酸激酶(PTEN-induced kinase 1,PINK1)在线粒体自噬中发挥主导作用,其积聚在功能失调的线粒体外膜上,进行自磷酸化后募集E3连接酶Parkin以泛素化线粒体外膜,通过与p62、LC3结合使溶酶体中蛋白水解酶释放降解线粒体,从而触发自噬。线粒体自噬的增加减少mtROS的生成、抑制NLRP3激活并减弱炎症损伤,而PINK1沉默或自噬抑制剂阻断自噬则逆转这一结果[30]。此外,HU等人[31]探究阻断自噬对高尿酸血症的影响发现,Beclin-1 siRNA转染抑制自噬,减少自噬溶酶体降解后组织蛋白酶B释放,从而抑制NLRP3活化和焦亡,从而减缓肾纤维化进程。综上可知,线粒体自噬是维持细胞稳态的重要生理过程,通过负调控炎症小体的活化减轻炎症反应损伤发挥关键作用,然而,所涉及自噬途径及自噬与NLRP3炎症小体之间的相互作用需要进一步研究。
2 NLRP3炎症小体与阿尔茨海默症
NLRP3炎症小体在AD中发挥重要作用。患者脑组织中NLRP3炎症小体的活化与β-淀粉样蛋白沉积、高度磷酸化tau蛋白导致线粒体功能障碍、裂解溶酶体等途径密切相关,其高表达诱发神经炎症加快疾病发展进程,而激活炎症小体加剧淀粉样斑块和神经纤维缠结病理过程,二者形成恶性循环从而共同导致认知功能障碍。
2.1 NLRP3炎症小体在阿尔茨海默症中表达升高
神经炎症是AD的病理学标志之一,与神经炎症密切相关的免疫细胞为小胶质细胞和星形胶质细胞。NLRP3炎症小体的组装和活化是神经炎症的重要组成部分,然而功能性炎症小体的形成和IL-1β分泌部分存在于先天免疫反应相关的小胶质细胞中,AD患者小胶质细胞中NF-KB核易位激活NLRP3炎症小体诱发神经炎症,从而加快认知功能障碍发展进程[32]。采集AD患者的外周血并提取单核细胞,深入分析炎症小体复合物中的基因和蛋白质发现,PYCARD(apoptosis speck-like protein containinga caspase activation and recruitment domain,PYCARD)、NLRP3、胱天蛋白酶1表达含量显著增加,下游细胞因子 IL-1β、IL-18 mRNA水平升高,提示NLRP3炎症小体在AD患者体内激活[33]。对中度和重度AD患者死后的脑组织颞叶皮层进行检测,与神经病理学未受影响的对照受试者相比,患者脑组织中NLRP3、ASC和胱天蛋白酶1蛋白质含量高于对照组,同时星形胶质细胞的活化标志物GFAP(glialfibrillaryacidicprotein,GFAP)基因表达显著升高[34]。在动物研究中,5XFAD( 5 familial AD mutations,5XFAD)双基因敲除AD小鼠模型的海马组织和大脑皮层内,调控细胞坏死性凋亡的胱天蛋白酶8表达升高,其一方面激活NF-κB信号通路介导炎性因子释放,另一方面增加ASC斑点生成调控NLRP3组装诱发炎症[35]。此外,细胞研究中,Wang等人以APP/PS20小鼠中LPS刺激提取的原发性小胶质细胞为研究对象,探究NLRP3炎症小体是否活化,发现NLRP3表达水平增加,而慢病毒介导的TREML2敲低则降低NLRP3含量,抑制炎症损伤神经元[36]。因此,综上可知AD患者脑组织、血浆中存在高水平IL-1β、IL-18,并且NLRP3蛋白表达增加,证明NLRP3炎症小体活化诱导炎症反应损伤神经元,从而加剧AD患者认知障碍程度。
2.2 NLRP3炎症小体与阿尔茨海默症的病理学联系
阿尔兹海默症患者大脑存在淀粉样蛋白斑块、神经原纤维缠结和神经炎症等病理特征。Aβ在神经元体内以淀粉样蛋白前体为底物通过β分泌酶和γ分泌酶水解生成,生理水平上Aβ调节突触可塑性、增强学习记忆能力、减少突触兴奋性毒性产生[37],而在AD患者中,Aβ集聚形成具有不同分子量和构象的Aβ低聚物,诱导线粒体功能障碍、溶酶体破裂等途径发挥神经毒性作用损伤认知功能[38]。此外,Aβ促进tau蛋白过度磷酸化降低其对微管的亲和力,从而形成细胞内聚集体加速认知衰退[39]。越来越多的研究发现,β-淀粉样蛋白沉积和过度磷酸化tau蛋白聚集与先天免疫激活密切相关,而且金属离子稳态与AD病理蛋白质之间存在密切关系[40]。Aβ聚集可激活小胶质细胞中NLRP3为主的炎性小体,而诱发的神经炎症又驱动Aβ聚集与tau蛋白过度磷酸化病理过程[41],二者相互影响共同导致神经变性。
2.2.1 Aβ、tau蛋白导致线粒体功能障碍介导炎症小体活化 在启动阶段,Aβ作为DAMPs与TLR4结合触发小胶质细胞活化,诱导NLRP3、pro-IL-1β转录,导致胱天蛋白酶1活化并促进IL-Ιβ、IL-18的成熟和分泌[42]。最新研究发现,线粒体功能障碍是NLRP3炎症小体激活的重要调节因子。一方面,Aβ通过调控小胶质细胞有氧糖酵解转变诱发高度促炎反应,潜在机制为Aβ激活脾酪氨酸激酶(spleen tyrosine kinase,Syk)并调控下游介导线粒体裂变/融合的调节因子AMPK,其失活后上调分裂蛋白Drp 1表达,线粒体过度裂解和电子传递链的破坏促进炎症小体NLRP3激活信号ROS的生成, 诱导NLRP3、 caspase-1表达升高从而驱动炎症反应[43],此外,Aβ在RAGE-TXNIP轴的作用下从小胶质细胞表面转运到线粒体,Aβ与Drp 1相互作用引发线粒体裂变导致线粒体功能障碍,从而激活NLRP3炎症小体[44]。另一方面,线粒体功能障碍导致线粒体mtDNA通过通透性转换孔释放至细胞质活化炎症小体[45]。有研究显示,NLRP3在AD中活化与Tau蛋白也相关,其过度磷酸化可增强Aβ神经毒性,而Aβ驱动tau磷酸化和集聚,二者协同靶向线粒体扰乱ATP生成,导致Ca2+流入胞内且线粒体过度摄取损伤其功能,从而调控炎症小体活化[39]。因此,AD患者脑组织中错误折叠蛋白集聚通过多途径损害线粒体功能从而活化NLRP3炎症小体。
2.2.2 Aβ、tau蛋白导致溶酶体破裂介导炎症小体活化 小胶质细胞募集斑块周围识别、吞噬Aβ,诱导溶酶体肿胀破坏其稳态,触发组织蛋白酶B释放激活NLRP3炎症小体[46]。AD的另一病理特征神经纤维缠结,由过度磷酸化Tau蛋白集聚形成。研究发现,Tau蛋白也被证实可通过小胶质细胞摄取、溶酶体去稳定化和组织蛋白酶B释放一系列途径激活NLRP3-ASC炎性体,增加IL-Ιβ分泌[47]。然而,NLRP3炎症小体的激活降低小胶质细胞吞噬能力,NLRP3抑制剂处理后胶质细胞吞噬Aβ能力增强,减少Aβ在脑组织中积累,从而抑制溶酶体裂解激活NLRP3炎症小体途径,揭示NLRP3炎症小体的活化促进Aβ沉积。此外,NLRP3炎症小体激活也干扰Tau蛋白的磷酸化。NLRP3调节Tau磷酸化所需磷酸酶和激酶活性增加Tau过度磷酸化,损伤空间记忆障碍,且其活化后释放IL-Ιβ诱导tau集聚加速神经元变性[41],证明NLRP3炎症小体的活化诱导Aβ、Tau蛋白通过正反馈通路促进AD的发展进程。
2.2.3 金属离子促进Aβ、tau蛋白集聚介导炎症小体活化 金属离子在中枢神经系统中参与神经元间信号传导,金属氧化态的多种酶催化神经肽生成,从而维持神经功能。研究发现,在AD病理学中,金属离子的稳态失衡损害神经网络,诱导氧化应激、突触损伤,此外,脑组织淀粉样斑块和神经纤维缠结中发现金属沉积,且过量的金属诱导小胶质细胞活化,促进炎症因子释放[48]。淀粉样蛋白前体参与大脑中铜、铁等金属离子的稳态调控,通过氧化Fe2+为Fe3+和还原Cu2+为Cu从而有利于从大脑清除,然而,氧化态金属离子可刺激β-分泌酶裂解淀粉样蛋白前体,并促进Aβ聚集从而降低低聚物溶解度,同样,与tau蛋白的较高结合亲和力促进神经纤维缠结形成[49]。另一方面,金属离子失衡诱导ROS过量生成,降低超氧化物歧化酶活性诱发氧化应激反应[50, 51]。ROS作为激活NLRP3炎症小体的信号分子,促进炎症小体活化诱导炎症级联加剧认知障碍。此外,有研究证明铁稳态破坏以激活NF-κB通路介导小胶质细胞释放IL-1β加剧促炎作用[52],然而,金属离子与NLRP3炎症小体具体作用关系尚不明确,但金属离子通过影响AD病理蛋白质激活NLRP3炎症小体得到广泛研究。
由此可见,Aβ聚集、Tau蛋白过度磷酸化通过多种途径参与NLRP3炎症小体活化,且介导神经炎症加剧AD患者淀粉样斑块和神经纤维缠结的形成,二者相互作用,扩大认知损伤效应(Fig.2)。因此,靶向抑制NLRP3炎症小体的活化可能是有效减缓AD发展的有效靶点。
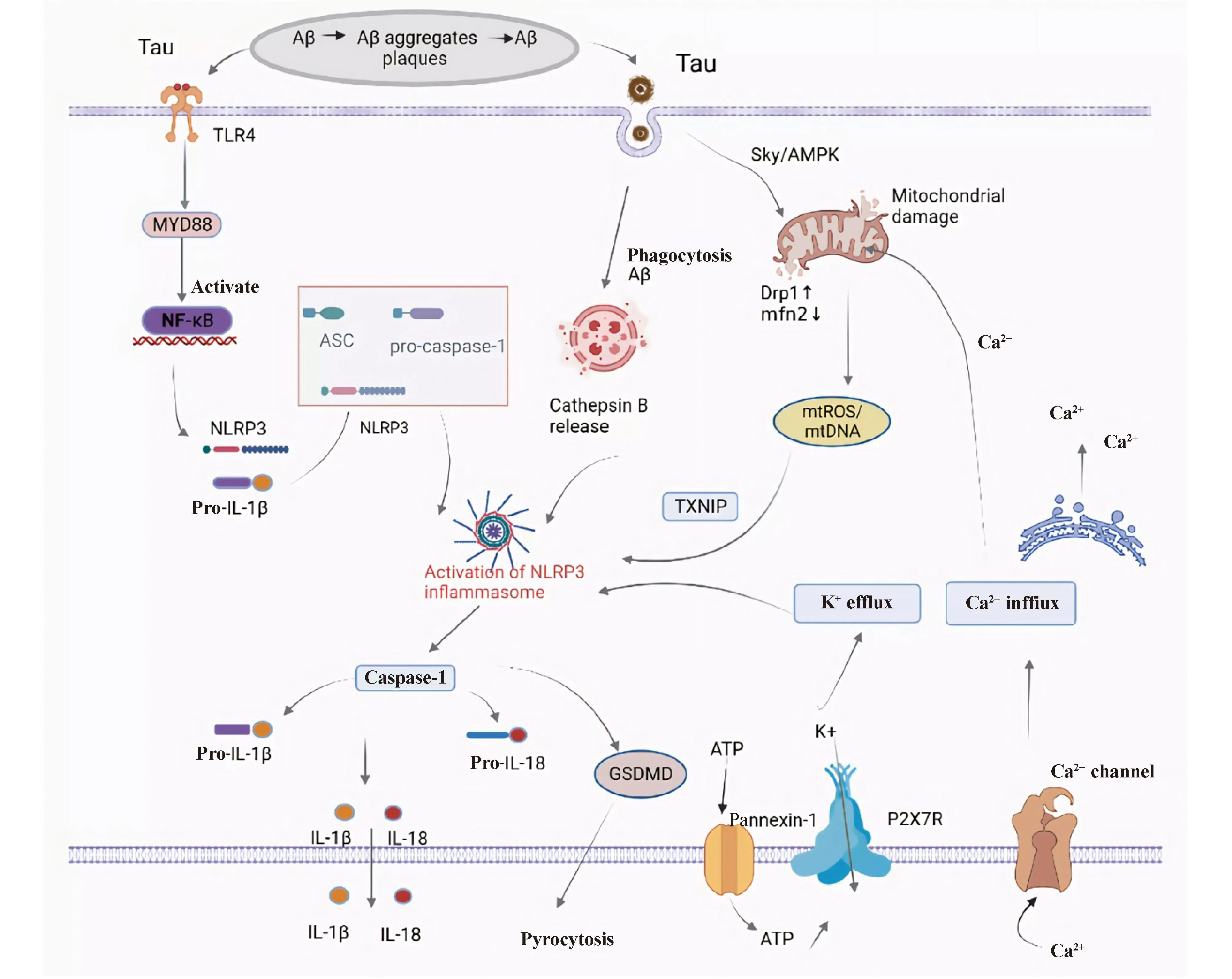
Fig.2 Activation mechanism of NLRP3 inflammasome in AD Firstly, protein aggregates (Aβ and tau) are recognized by microglial pattern recognition receptors,such as Toll-like receptors (e.g., TLR4),leading to activation of the MYD88/NF-κB pathway. The activation of the NF-κB pathway promotes a signaling cascade, resulting in the transcription of pro-IL-1β and NLRP3. NLRP3, ASC, and pro-caspase-1 assemble together to form the NLRP3 inflammasome, which subsequently activates caspase-1, then cleaves pro-IL-1β to produce the active form of IL-1β and secretes it extracellularly. Multiple molecular orcellular events, including ionic flux, mitochondrial dysfunction and reactive oxygen species (ROS)generation, and lysosomal damage, have been shown to activate the NLRP3 inflammasome
3 靶向NLRP3炎症小体改善阿尔茨海默症的治疗策略
鉴于NLRP3炎症小体在AD发病机制中的关键作用,探索以 NLRP3 炎症小体为靶点的AD治疗策略也逐渐成为该领域研究热点。迄今为止,研究表明多种药物及治疗措施可直接靶向NLRP3蛋白或炎症小体成分抑制其活化改善AD。
3.1 靶向炎症小体的抑制剂及化合物药物治疗
大量研究发现,部分小分子抑制剂和化合物靶向NLRP3炎症小体治疗AD发挥不可忽视的作用,其通过调控AKT/MAPK、ROS/TXNIP/NLRP3、AMPK/PINKI/Parkin等通路介导氧化应激、自噬途径抑制NLRP3炎症小体活化改善AD认知障碍。
MCC950是NLRP3炎症小体的选择性抑制剂,通过靶向Walker B基序内部或附近的炎症小体位点,阻断ATP水解,影响Walker B功能使NLRP3的活性构象关闭从而阻止NLRP3激活[53]。Dempsey等[54]以APP/PS1 AD小鼠模型为研究对象,发现 MCC950可通过刺激Aβ吞噬,减少Aβ的异常集聚,从而抑制炎症小体活化、减少caspase-1的成熟释放。JC-124是可以渗透血脑屏障的抑制剂,可减轻AD相关的Aβ沉积、氧化应激等病理缺陷,其潜在机制为抑制ASC聚集和IL-1β分泌,并且降低β-分泌酶活性减少Aβ生成,同时星形胶质细胞的增生增强Aβ吞噬作用,二者共同降解淀粉样斑块,另一方面,因突触可塑性对IL-1β敏感,JC-124处理后IL-1β分泌减少且增加突触素表达,更好的修复突触可塑性,减缓AD患者认知能力下降进程[55]。靶向炎症小体的抑制剂OLT1177抑制NLRP3激活后,皮层斑块数量减少,谷胱甘肽过氧化物酶含量升高减弱氧化应激,揭示OLT1177可改善空间学习记忆能力[6]。此外,Aβ聚集体和NLRP3炎症小体协同作用增加神经元损伤,BPBA靶向二者联合治疗增强疗效,研究证实BPBA抑制Cu2+或zn2+诱导Aβ聚集,降低Aβ聚集体的神经毒性,同时通过减少炎症小体适配器蛋白质ASC形成,抑制炎症小体下游信号caspase-1的激活从而缓解认知障碍[56]。NLRP3炎症小体的活化与ROS的异常积累相关,ROS可能通过NF-κB信号通路调控NLRP3炎性小体的启动,也可能激活caspase-1释放促炎因子IL-18、IL-1β,多种抗氧化化合物可调控ROS/NLRP3/IL-1β轴抑制神经炎症。艾地苯醌是线粒体电子传递链中电子供体辅酶Q10的类似物,通过维持线粒体膜电位稳定抑制ROS的生成和NF-kB/STAT3信号传导,从而抑制NLRP3炎症小体活化[57]。TXNIP是连接炎症和氧化应激的桥梁,ROS导致TXNIP与TrX解离后与NLRP3相互作用促进炎症小体形成激活,而Wang等[58]以DI-NBP干预AD小鼠模型发现,干预组小鼠大脑中TrX活性增强,TXNIPmRNA水平显著降低,抑制TXNIP与NLRP3相互作用,并且降低ASC表达,从而减轻炎症和神经退行性变。研究还发现,褪黑素、金香酰胺、β-羟基丁酸、CSB6B等化合物均可抑制炎症小体活化,具体机制见Table 1。
3.2 中药治疗
在AD的基础研究中,多种中草药提取物已被证明可靶向NLRP3炎症小体减轻神经炎症损伤,为AD的临床治疗提供理论依据。
猴头菌具有抗氧化活性。在注射AICI3诱导的AD模型中,猴头菌通过上调大鼠海马组织中Nrf2表达,增强SOD和 GSH活性从而抑制NLRP3炎症小体活化,减少海马神经元变性和改善大鼠行为[73]。此外,中药配方芍药甘草汤也可通过介导氧化应激抑制神经炎症,最终修复神经突触可塑性。
芒果苷是一种从植株中提取的多酚,研究证明其具有抗氧化、抗炎、抗菌等特性。Yan等[74]以LPS刺激构建BV2细胞模型为对象研究发现,其通过抑制NF-κB激活,下调NLRP3表达,降低促炎介质iNOS、IL-1β转录,从而改善AD患者学习记忆功能[75]。
红景天苷是从红景天玫瑰中提取的主要活性成分。Cai等[76]证明红景天苷抑制NLRP3炎症小体介导的焦亡保护神经元,其潜在机制为通过调节TLR4/ MyD88 /NF-κB信号通路,抑制ASC、cleaved Caspase-1表达,并减少Aβ和Tau蛋白聚集,最终减少海马神经元损伤。此外,黄芩苷也被证实可通过调控TLR4/NF-κB信号促进认知功能恢复[77]。
荔枝籽多酚通过AMPK通路激活自噬,促进错误折叠蛋白质聚集体的吞噬和降解,从而抑制NLRP3炎症小体过度活化,减轻APP/PS1小鼠海马区神经元损伤[78]。
3.3 其他
除了NLRP3炎症小体抑制剂及化合物、中草药有效改善AD发展进程外,一些中医技术和康复方法可抑制NLRP3炎症小体活化减轻神经炎症损伤。经研究表明,空间训练可缓解AD的记忆丧失,对PR5小鼠进行连续四周水迷宫和新目标识别测试训练,可显著提高其认知能力,增加海马突触素含量,同时逆转PR5小鼠体内的高NLRP3、caspase-1和IL-1β的表达水平[79]。Rosa等[80]以脑室注射Aβ1-40诱导的AD模型为研究对象探究中度跑步机运动对小鼠海马NLRP3表达影响,发现体育锻炼可抑制ROS诱导的氧化应激,降低TXNIP的免疫含量,降低NLRP3、caspase-1/procaspase-1的比例,改善小鼠的体育锻炼能力。另外,中医传统技术电针疗法也可改善模型小鼠的记忆障碍,Lin等[81]通过电针联合治疗5xFAD小鼠的GB13本神穴和GV24神庭穴,可上调AMPK和AKT激酶活性,激活自噬溶酶体途径的主要转录因子Tfeb,促进NLRP3炎症小体自噬降解,抑制炎症小体活化。
4 问题与展望
AD是一种功能和认知能力下降为特征的综合征,越来越多的证据表明NLRP3炎症小体在AD的病理机制中起着不可或缺的作用,目前为止,许多炎症小体抑制剂、化合物和中草药提取物及其他治疗措施在体外和多种AD模型中可有效抑制NLRP3炎症小体活化、减少神经元死亡和修复突触可塑性,但转化为临床应用和临床疗效、安全性仍需进一步验证。有报道显示,MCC950、OLT1177、CY-09等抑制剂已进行临床实践,也显示出相对较高的安全性,特别强调的是,NLRP3炎症小体抑制剂可直接作用于NLRP3本身,也可作用于NLRP3炎性体活化的上下游因子,例如:NEK7、ASC、caspase-1和IL-1β,揭示靶向NLRP3炎症小体对AD的治疗具有广阔的治疗前景,但这需要未来利用调控机制开展大规模临床试验。其次,一些中草药具有一定的毒性,其提取物在维持药物活性和降低不良反应前提下,避免产生神经毒性是当前需要解决的问题,再者,靶向NLRP3炎症小体的化合物是否通过血脑屏障和通过血脑屏障的途径需进一步阐明。此外,离子稳态、溶酶体和线粒体功能破坏等触发NLRP3炎症小体活化及他们如何相互关联并聚焦于AD中的NLRP3活化机制仍待进一步深入研究,并且AD病理标志物Aβ、Tau蛋白与NLRP3炎性小体相互影响机制尚未完全明确。因此,为了推动靶向NLRP3炎症小体的治疗应用于临床,深入阐明NLRP3炎症小体在AD中的作用机制,探索以NLRP3炎症小体为靶点的AD治疗策略,开发具有安全性和较小不良反应的药物减缓认知障碍是未来研究方向,其为AD临床治疗提供新思路与策略。
