抗松材线虫病马尾松种质资源遗传多样性分析及核心种质构建
2024-02-25邓莉丽刘青华周志春骆定会
邓莉丽,刘青华,周志春,高 凯,骆定会
(1.中国林业科学研究院 亚热带林业研究所 浙江省林木育种技术研究重点实验室,浙江 杭州 311400;2.南京林业大学 林学院,江苏 南京 210037;3.浙江省临海市自然资源和规划局,浙江 临海 317000)
松材线虫Bursaphelenchusxylophilus病是20 世纪80 年代初传入中国的一种毁灭性森林病害,其传播速度快,防治难度大,危害中国松林面积超180 万 hm2,其中,马尾松Pinusmassoniana最易受感染,松材线虫病对中国马尾松林业经济发展造成了极大破坏[1]。马尾松具有速生、耐旱等优良特点,松林面积达804 万 hm2,是中国南方荒山造林的先锋树种[2]。近40 a 的林业生产实践和研究发现,松树种间和种内不同个体对松材线虫病的抗性存在较大差异,且马尾松群体中存在抗病基因型,因此选育抗性品种是当前治理松材线虫病害最经济有效的途径之一[3]。自2001 年开始,中国开展了马尾松松材线虫病抗性育种研究,现已收集保存一批抗松材线虫病马尾松种质资源[4-6]。种质资源是遗传改良的物质基础,种质资源收集保存的越多,其群体遗传多样性越高,但收集保存的成本也越高。FRANKEL 等[7]提出核心种质的概念,即用最小的遗传资源数量和最小的遗传冗余最大限度地代表整个遗传资源的多样性。通过构建核心种质,能最大限度地去除重复和遗传关系较近的种质材料,加强现有抗性马尾松种质资源的有效保护利用,降低管理成本[8]。分子标记法是林木遗传多样性分析、种质资源评价和核心种质库构建常用的方法[9-13]。其中简单序列重复(SSR)分子标记具有多等位基因、共显性和高度多态性等优点,能够直观反映不同种质间遗传信息差异,在遗传育种中得到广泛应用[14]。LÜ等[15]利用 SSR 分子标记数据研究了桉树Eucalyptuscloeziana的遗传多样性并构建了核心种质;唐玉娟等[16]利用12 对 SSR 引物对杧果Mangiferaindica种质资源的遗传多样性进行分析并构建了分子身份证;HU 等[17]利用 SSR 分子标记技术对猕猴桃Actinidiachinensis的遗传多样性进行研究,构建了猕猴桃核心种质。
本研究利用 SSR 分子标记技术对前期筛选获得的103 份抗松材线虫病马尾松种质和安徽省林业科学研究院审定的高抗良种进行遗传多样性及亲缘关系分析,并基于M 策略和随机取样策略构建核心种质,通过分析不同构建策略的核心种质确定抗性马尾松核心种质的最适取样比例,为抗性马尾松种质资源的合理开发和利用以及挖掘优异基因提供理论支撑。
1 材料与方法
1.1 材料
2018 年,在国家马尾松种质资源库中依据松脂和抗性的关系,从1 207 株树中选择120 份候选抗性马尾松无性系,同年在松材线虫病重灾区严重感染林分中收集健康树242 份,并将所有无性系嫁接保存于浙江省临海市林业技术推广和场圃旅游服务总站(28°88′N,121°01′E)。2018—2020 年连续3 a 高强度对收集区内的候选抗性马尾松无性系进行人工接种松材线虫测定,筛选存活健康株系,共获得103 份抗性马尾松无性系,另取安徽省林业科学研究院审定的抗松材线虫病良种(为目前中国所有的抗松材线虫病良种)作为对照。合计114 份,其中浙江37 份、江西26 份、安徽42 份、福建6 份、贵州1 份、广西2 份。采集抗性样株幼嫩针叶,在-80 ℃冰箱中保存,各试验材料编号及来源见表1。

表1 材料编号及来源Table 1 Test materials’ number and source
1.2 方法
1.2.1 DNA 提取及SSR 引物信息 马尾松针叶基因组DNA 采用北京艾德莱生物科技有限公司生产的改良CTAB 植物基因组DNA 快速提取试剂盒提取。利用超微量分光光度计,根据在260 nm 的吸光度D(260)和D(260)/D(280)确定DNA 样品的纯度和质量浓度,经检验合格后的DNA 用TE 溶液或去离子水(灭菌后)稀释至20~50 mg·L-1,置于-20 ℃的冰箱保存备用。
参考董虹妤等[18]和YANG 等[19]使用的马尾松 SSR 引物,用质量浓度为1.2%的琼脂糖凝胶电泳检测,筛选条带清晰且多态性较高的SSR 引物(图1),最终筛选出具有高多态性引物14 对(表2),引物序列由浙江尚亚生物技术有限公司合成。
图1 引物多态性检测图Figure 1 Primer polymorphism detection diagram

表2 14 对SSR 引物信息Table 2 14 pairs of SSR primer information
1.2.2 SSR-PCR 扩增和毛细管电泳 利用选取的14 对 SSR 引物对114 份马尾松DNA 样本进行PCR 扩增,设定本研究的反应体系为25.0 μL:2×TaqPlus Master Mix (Nazyme Code:P211-03)12.5 μL,引物F 和R (10 μmol·L-1)各1.0 μL,DNA 模板2.0 μL,ddH2O 8.5 μL。PCR 扩增反应在Takara PCR Thermal cycler 上进行,其程序为:95 ℃预变性3 min;95 ℃变性15 s,60 ℃退火15 s,72 ℃延伸30 s,35 个循环;72 ℃延伸5 min,4 ℃保存。所得PCR 产物采用Qsep100 全自动毛细管电泳核酸分析仪进行检测和基因分型。
1.2.3 遗传参数及群体结构分析 利用GenAlex 6.5[20]计算各位点的遗传多样性参数,包括等位基因数(Na)、有效等位基因数(Ne)、观测杂合度(Ho)、期望杂合度(He)、Shannon’s 多样性指数(I)、近交系数(Fis)、固定指数(F)和基因流(Nm),并对收集的抗松材线虫病马尾松种质进行主坐标分析(PCoA)。采用Cervus 2.0[21]获得微卫星位点的多态信息含量(PIC)。利用Structure 2.3.4[22]对抗性马尾松种质资源进行分类,推测最佳聚类组群数目,确定群体结构,并对各亚群的遗传多样性参数进行分子方差分析(AMOVA)。
1.2.4 核心种质构建 对抗松材线虫病马尾松种质进行群体结构分析后,利用Power Core[23]和Core Finder[24]对各亚群基于不同算法构建核心种质。其中,Power Core 是利用随机取样策略和启发式算法,寻找从初始阶段到最终阶段的最优路径进行核心集合选择;Core Finder 基于NP-完全覆盖问题,采用拉斯维加斯式(LasVegas-style)随机算法的M 策略进行样本选择。在构建核心种质过程中,Power Core 和Core Finder 会自动生成合理的取样比例。最后利用SPSS 26.0 和Excel 中t检验对原有种质与核心种质各遗传多样性指标进行差异显著性分析,同时运用PCoA 和Powermarker[25]对所构建的核心种质构建UPGMA 进化树,确认构建的核心种质的代表性。
2 结果与分析
2.1 抗松材线虫病马尾松种质资源遗传多样性分析
由表3 可知:14 对 SSR 引物在114 份抗松材线虫病马尾松样本中共检测出115 个等位位点,Na为5~11,平均为8.17。所有材料的Ne为3.45~8.05,平均为5.54。Ho和He的分别为0~0.57 和0.51~0.73,He均值(0.64)高于Ho均值(0.06),Fis平均为0.92,综合表明该抗性马尾松种质中存在杂合子缺失现象。PIC为0.82~0.95,平均为0.9,I为1.15~1.90,平均为1.51,表明所选引物多态性高。

表3 不同SSR 位点的遗传多样性统计Table 3 Genetic diversity of different SSR microsatellite loci
2.2 抗松材线虫病马尾松种质群体结构和主坐标分析
Structure 分析得出的结果经Structure Haevester 处理后发现(图2A):lnP(D)(马尔科夫链不作数迭代后所得的对数似然值)持续增大,没有拐点,可用ΔK[L(K)在连续K之间变化率的增量]来确定最优分组数(K)。由图2B 可知:当K=4 时,ΔK取得最大值,表明参试的114 份马尾松样本被划分为4 个亚群。亚群Ⅰ(绿色)包含了12 份种质资源,其中2 份来自广西,1 份来自贵州,3 份来自福建,2 份来自安徽,4 份来自浙江;亚群Ⅱ(黄色)包含24 份种质资源,其中8 份来自安徽,7 份来自江西,9 份来自浙江,该亚群有12 份抗性马尾松种质掺杂有来自红色部分的基因;亚群Ⅲ(红色)包含了29 份种质资源,其中10 份来自安徽(均为皖马抗良种),8 份来自江西,11 份来自浙江,共有12 份抗性马尾松种质掺杂有来自黄色部分的基因;亚群Ⅳ(蓝色)包含49 份种质资源,其中2 份来自福建,21 份来自安徽(其中1 份为皖马抗良种),11 份来自江西,15 份来自浙江(图3)。由Structure 输出的分组结果可知(表4):筛选获得的抗性马尾松种质中有75.4%的个体样本隶属不同亚群的比例(Q)>0.8,仅有12.3%的个体Q<0.6,表明抗性马尾松种质群体结构较强,不同来源的个体被聚在相同的遗传结构中表明不同省间的马尾松存在基因渗入现象。
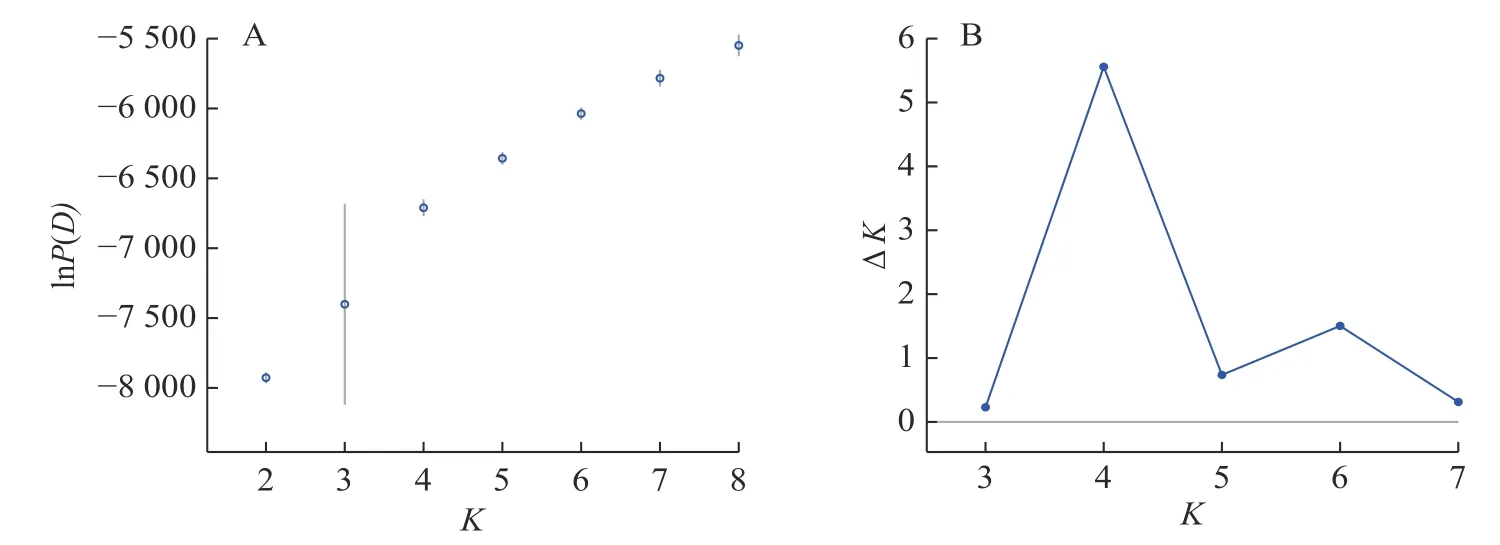
图2 lnP(D)和ΔK 随K 变化的折线图Figure 2 Line plots of lnP(D) and ΔK as a function of K
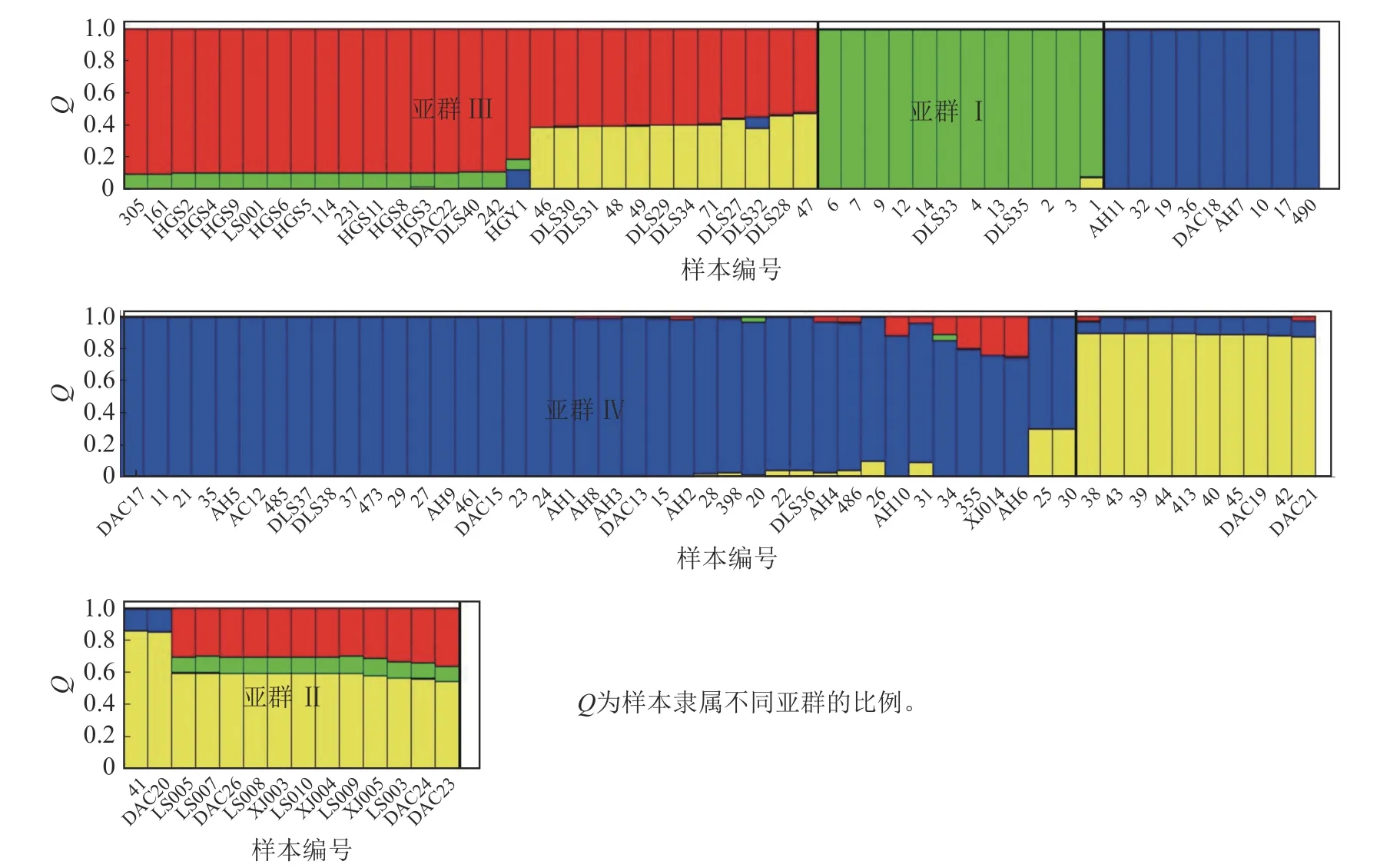
图3 114 份抗性马尾松样本的Structure 聚类图Figure 3 Structure cluster diagram of 114 samples of resistant P.massoniana

表4 各亚群基因型情况Table 4 Number of genotypes in each cluster
基于遗传距离矩阵对抗性马尾松材料进行主坐标分析(PCoA)。由图4 可知:全部马尾松种质资源可大致分为3 组,主坐标1 和2 分别解释了位点信息数据中10.45%和6.74%的变异。基于抗性马尾松114 份样本主坐标分析图可知:大部分地理来源一致的抗性马尾松种质分布比较集中,但来自安徽、江西、浙江的部分抗性马尾松种质在3 个组中均有分布。结合群体结构分析结果可知:114 份抗性马尾松种质资源的分布情况基本符合群体结构分析。
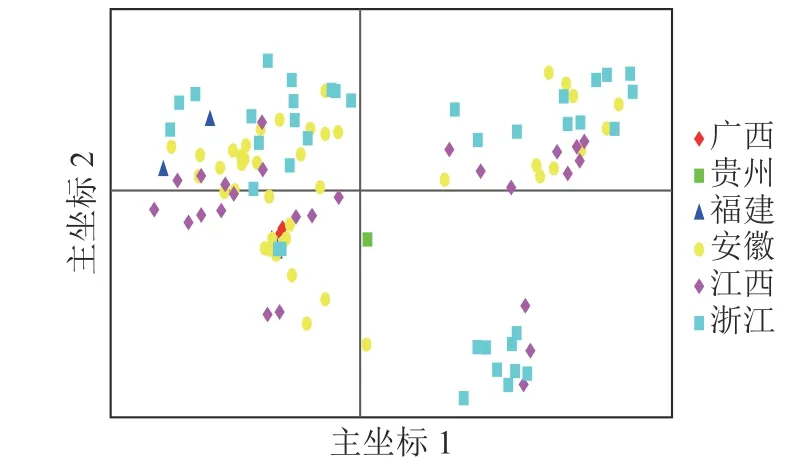
图4 抗性马尾松114 份样本主坐标分析Figure 4 Principal coordinate analysis of 114 samples
2.3 抗松材线虫病马尾松各亚群间遗传多样性及分子方差分析
由表5 可见:抗性马尾松种质各亚群的遗传多样性整体水平较高。亚群Ⅳ的等位基因数在4 个亚群中最大(13.43),亚群Ⅰ具有最低的等位基因数(6.07)。抗性马尾松4 个亚群所包含的马尾松个体数不同,但亚群间遗传多样性水平差异不明显。亚群Ⅳ的Ne最高,为7.14,而最低的是亚群Ⅰ,为4.87。亚群Ⅳ的Ho和He均最高,但与具有最小值的亚群Ⅲ差异不显著,分别高0.03 和0.15。
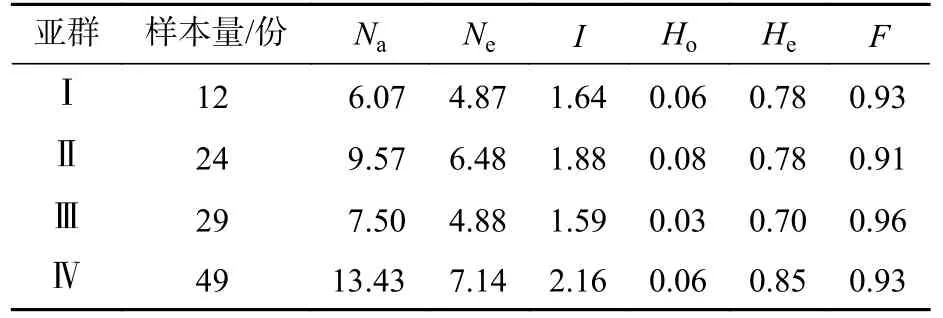
表5 马尾松各亚群遗传多样性水平Table 5 Genetic diversity parameters for all populations of resistant P.massoniana
AMOVA 方差分析表明(表6):抗性马尾松遗传变异14%存在于亚群间,而亚群内变异为80%,表明亚群内的遗传变异是抗性马尾松变异的主要来源。4 个亚群间FST为0.143,表明亚群间存在中等程度遗传分化。

表6 基于微卫星数据的遗传变异分析Table 6 Analysis of molecular variance from microsatellite data using GenALEx
2.4 抗松材线虫病马尾松核心种质库构建与评价
利用M 策略和随机取样策略构建核心种质的遗传多样性指标见表7。结果显示:基于随机取样策略的Power Core 和基于M 策略的Core Finder 分别筛选得到的核心种质均保留了原有种质100%的等位基因数。但Core Finder 核心种质包含的种质(72 份)少于Power Core (79 份),且两者间各遗传多样性参数差异不显著(P>0.05)。综合以上分析表明:选择M 策略抽取72 份抗性马尾松种质能够以最小的种质份数最大程度地代表收集区内抗性马尾松种质资源的遗传多样性。利用M 策略构建的核心种质中包含广西2 份(100.0%)、贵州1 份(100.0%)、福建4 份(66.7%)、安徽33 份(其中4 份为皖马抗良种)(78.6%)、江西8 份(30.8%)和浙江24 份(64.8%)种质材料。

表7 不同策略构建的核心种质遗传多样性指标比较Table 7 Comparisons of genetic diversity index of core collections constructed by different tactics
由表8 可见:构建的72 份抗性马尾松核心种质占114 份原有种质的63.16%,其Na均值、Ne均值、I均值分别占原有种质相应遗传参数的100.00%、95.67%和94.96%。t检验结果表明:列入研究的抗性马尾松种质与入选的核心种质的遗传多样性无显著差异(P>0.05),说明本研究基于M 策略构建的抗性马尾松核心种质能充分代表列入研究的原抗性马尾松种质资源的遗传多样性,剔除了与114 份原有种质重复或亲缘关系较近的材料。

表8 72 份抗性马尾松核心种质与114 份原有种质遗传多样性对比Table 8 Comparison of genetic diversity between 72 core germplasm and 114 original germplasm of resistant P.massoniana
PCoA 分析(图5)显示:抗性马尾松核心种质均匀分布在整个马尾松种质资源中,表明本研究所构建的抗性马尾松核心种质具有很好的代表性。UPGMA 进化树(图6)显示:72 份抗性马尾松可分为3 个类群,其中类群Ⅰ的11 份抗性马尾松种质全部位于亚群Ⅰ(91.7%),类群Ⅱ的30 份抗性马尾松种质全部位于亚群Ⅳ(61.2%),类群Ⅲ的抗性马尾松种质18 份位于亚群Ⅱ(75.0%),其余13 份位于亚群Ⅲ(44.8%)。结合结构分析可知:位于亚群Ⅱ中混杂有黄色和红色的核心种质与亚群Ⅲ中的核心种质聚为一个亚类,亚群Ⅱ中以黄色为主的核心种质聚为另一个亚类,但抗性马尾松核心种质的UPGMA 聚类结果与列入研究的原有种质结构分析和PCoA 分析结果基本一致,进一步对核心种质进行了确认。

图5 72 份核心种质与114 份原有种质主坐标分析Figure 5 Principal coordinate analysis of 72 core germplasm and 114 original germplasm

图6 72 份抗性马尾松核心种质的UPGMA 聚类图Figure 6 UPGMA clustering map of 72 samples of resistant P.massoniana
3 讨论
3.1 抗松材线虫病马尾松种质资源遗传多样性及群体结构
遗传多样性在生物育种工作中至关重要,是评价生物多样性的重要组成部分,物种的遗传多样性越高或遗传变异越丰富,其对环境变化的适应能力也就越强[26]。本研究利用14 对 SSR 引物对经高强度人工接种松材线虫后存活下来的103 份马尾松种质和安徽省林业科学研究院审定的11 份高抗良种进行遗传多样性和群体结构分析。Na是衡量SSR 位点多态性高低的重要指标,其中所有种质的平均Na为8.17,高于沈敬理等[27]利用SSR 分子标记技术对131 个马尾松无性系的研究(Na=3.67),可能是本研究选择的 SSR 引物具有较高多态性和基因分型检测手段不一样造成的。Ne、He、I等是评价遗传多样性的重要指标。高景斌等[28]利用 SCAR 分子标记对抗性马尾松材料进行遗传多样性研究,其Ne和I分别为1.51 和0.46;YANG 等[19]运用20 对 SSR 引物分析马尾松二代亲本种质的遗传多样性,其Ne为3.01,I为1.26,PIC为0.89;董虹妤等[18]对选自马尾松第2 代育种群体的12 份马尾松材料运用13 对 SSR 引物进行研究,其中Na和I分别为3.17 和0.47。与上述研究结果相比,本研究所选择的14 对 SSR 引物能有效用于检测马尾松的遗传多样性(Ne=5.54、He=0.64、I=1.51、PIC=0.90),同时表明筛选获得的103 份抗性马尾松种质和安徽省林业科学研究院审定的11 份良种具有丰富的遗传多样性,在未来的抗性育种进程中具有很大的遗传增益潜力。
Structure 结构分析、UPGMA 聚类分析和PCoA 分析被广泛应用于推断植物的群体结构,但三者的统计原理不同。PCoA 分析是一种非约束性的数据降维方法,通过对特征值和特征向量进行排序来寻找主坐标,从而有效地绘制所分析的样本;Structure 结构分析基于贝叶斯模型的计算方法,根据基因型信息对多组样本进行分类;UPGMA 聚类分析利用非加权组平均法对参试样本进行层次聚类。PCoA 和UPGMA 聚类分析可直观看到材料间的分类关系,但Structure 结构分析能清晰地反映材料间的遗传背景和基因交流情况[29-30]。总体而言,Structure 结构分析、UPGMA 聚类分析和PCoA 分析相辅相成,两两使用有助于全面了解不同样本间的遗传差异。本研究Structure 结构分析结果表明:114 份抗性马尾松种质资源被分成4 个亚群,而PCoA 分析将所有抗性马尾松种质划分为3 组,将结构分析所得4 个亚群中的亚群Ⅱ和亚群Ⅲ合并为一组,可能是因为这2 个亚群中的部分抗性马尾松种质Q<0.6,存在一定频率的基因交流和渗透。在结构分析中,每个亚群内都包含来自不同地区的个体,地理来源一致的抗性马尾松种质并没有完全聚在一起,这可能是由于不同地区育种者之间亲本交换或频繁的人工引种,导致新环境中种质的遗传信息发生了改变。安徽省林业科学研究院审定的11 份抗松材线虫病良种是中国所有的抗松材线虫病马尾松优良种质,已在安徽省松材线虫病疫区进行了大面积的林相改造和造林,在松材线虫病防控中发挥了重要作用。这11 份抗松材线虫病良种主要分布在亚群Ⅲ中,仅与19 份抗性马尾松种质聚为一个亚群,这表明本研究筛选获得的大部分抗性马尾松优良品系与审定的良种遗传关系较远,具有较低的遗传相似性。因此,本研究筛选获得的103 份抗性马尾松种质与审定的11 份良种遗传信息同质性程度较低,且具有较高的遗传多样性,可保障今后营造的抗性林在生态系统上具有较强的稳定性。
3.2 抗松材线虫病马尾松核心种质库构建
传统的核心种质构建方法是对比不同种质资源在植物学、形态学等性状上的差异来剔除亲缘关系相近的样本并且抽取核心种质,但表型性状容易受到外界因素影响而表现不稳定[31]。基于分子标记数据构建核心种质可减少温度、气候、环境等外界因素的影响,能够很好地作为植物形态和生理特性的有效补充。本研究根据SSR 分子标记基因分型数据,利用Core Finder 和Power Core,基于M 策略和随机取样策略构建抗松材线虫病马尾松核心种质。综合对比分析不同构建策略,得到72 份抗性马尾松核心种质,其保留了列入研究的原有种质全部的等位基因数,且各遗传多样性参数如Ne、I、He、PIC分别为5.30、1.43、0.63、0.92,均与114 份原有种质的遗传多样性参数无显著差异(P>0.05)。本研究构建的72 份抗性马尾松核心种质在广西、贵州、福建、安徽、江西和浙江6 省中均有分布,但江西抗性马尾松核心种质抽取比例为30.8%,低于其他省份抗性马尾松种质的入选比例,表明该省抗性马尾松样本存在较高的遗传相似性。结合结构分析可知:72 份抗性马尾松核心种质全面涵盖了列入研究的抗性马尾松种质的4 个亚群,亚群Ⅲ抗性马尾松核心种质入选比例最低(44.8%),表明该亚群抗性马尾松种质遗传信息比较一致;亚群Ⅰ的抗性马尾松核心种质入选比例最高(91.7%),表明该亚群可能存在最丰富的遗传多样性。
取样比例的大小是核心种质库能否构建成功的重要因素。WANG 等[32]和ESCEIBANO 等[33]认为核心种质取样比例为5%~40%,大多数为5%~15%。李魁鹏等[34]利用SSR 分子标记从864 份杉木Cunninghamialanceolata中筛选获得了80 份核心种质,占原有种质的9.3%;黄小凤等[35]利用SNP 分子标记从221 份荔枝Litchichinensis品种(品系)中获得了44 份核心种质,占原有种质的20%;张馨芳等[36]基于SSR 分子标记数据,从161 份板栗Yanshanchestnut中构建了含46 份种质的核心种质,取样比例为28.6%。但本研究核心种质取样比例为63.13%,高于其他木本植物的取样比例。李自超等[37]认为核心种质取样比例通常由原有种质的数量、群体结构和遗传多样性水平等决定,对于遗传多样性丰富、原有种质数量较小的群体,可适当增大取样比例。
4 结论
利用14 对 SSR 引物对114 份抗松材线虫病马尾松种质的遗传多样性分析表明:所选抗性马尾松种质具有丰富的遗传多样性(Na=8.17,Ne=5.54,He=0.64,I=1.51,PIC=0.90),且本研究筛选的103 份抗性马尾松材料遗传结构简单、遗传变异丰富,可用于开展马尾松重要性状的关联分析研究。根据SSR 分子标记数据基于M 策略构建了含有72 份材料的抗性马尾松核心种质,保留了列入研究的原有种质100%的等位基因。经过t检验、PCoA 分析和UPGMA 聚类分析,构建的抗性马尾松核心种质与列入研究的原有种质之间无显著差异,剔除了遗传相近的材料,具有良好的代表性。
