限制性内切酶酶切后去磷酸化联合蓝白斑筛选检测KRAS基因第12位稀有突变
2024-02-24周翠兰陈婕许云思付乙人彭翠英
周翠兰, 陈婕, 许云思, 付乙人, 彭翠英
南华大学衡阳医学院 1.基础医学院应用解剖学与生殖医学研究所, 2.生态健康与人类重要疾病防控湖南省高校重点实验室,3.生物毒理与生态修复衡阳市重点实验室, 4.有色金属矿区耕地重金属污染生态阻抗技术研究所衡阳市重点实验室,湖南衡阳 421001
KRAS基因突变与多种肿瘤的发生发展相关,据COSMIC体细胞突变数据库,绝大多数的KRAS基因突变为点突变,较常见的为第12位、第13位密码子突变,其野生型序列为TGGTGG,其中第12位突变具有较高的发病频率,最常见的突变类型为TGATGG与TGGTGA[1-2]。蓝白斑筛选原理为,外源性DNA片段与含lacZ的载体连接,插入到其多克隆位点,破坏lacZ的阅读框架,导入宿主为缺陷菌株,致β-半乳糖苷酶基因断裂而不能产生分解培养基中的X-gal的β-半乳糖苷酶,所以克隆颜色呈白色[3-4]。研究发现,含终止密码子的外源性DNA片段(长度为3的倍数)插入到含lacZ的载体中,克隆颜色呈白色;不含终止密码子的外源性核苷酸片段(长度为3的倍数)插入到含lacZ的载体中,呈蓝色克隆[4]。
本课题组利用蓝白斑筛选技术已完成了KRAS基因第12位密码子突变检测,可检测出千分之一的稀有突变[5],但不能满足肿瘤早期临床诊断的需要。为降低突变检测的下限,本研究采用限制性内切酶酶切后去磷酸化联合蓝白斑筛选对KRAS基因第12位密码子突变进行检测,为临床肿瘤早期筛查提供理论与技术支持。
1 材料和方法
1.1 主要仪器与试剂
PCR仪(A200型)购自中国杭州朗基公司,E Coli.DH5α菌株、100 bp DNA Ladder、PMD-19T购自中国北京天根公司。dNTP、BstNI、EcoRI、KpnI、T4连接酶购自美国NEB公司。质粒DNA提取试剂盒购自中国大连TaKaLa公司。MixTaq与DNA Polymerase购自美国Thermo Scientific公司。IPTG、X-gal、Amp、Tris-base试剂购自中国长沙宏宇生物公司。人工模板KRAS基因第12位密码子突变型(mutant type,MUT)与野生型(wild type,WT)质粒为本实验室构建,插入长度为102 bp(3的倍数)的核苷酸片段,第12位密码子MUT 12质粒其TGG突变成TGA。
1.2 引物设计与合成
根据COSMIC体细胞突变数据库显示的KRAS基因第12位密码子热点突变,NCBI网站(https://www.ncbi.nlm.nih.gov/pubmed)显示的KRAS基因序列,利用引物设计软件Premier 5.0设计引物,设计引物主要考虑成功引入酶切位点,将引入突变的碱基置于引物的3′末端。设计一对引物:FS(正向引物)、AS(反向引物)。FS中3′的C碱基为引入BstNI酶切序列的C碱基。FS与AS扩增得到的产物双酶切后用于亚克隆,双酶切后获得的插入片段长度为102 bp,插入片段的长度为3的倍数。AS的序列为5′-CTACGGAATTCTTGGACCATATTCGTCCACA-3′,FS的序列为5′-ATACGAGGGTACCTGACTGCATACAAACTTGTGGTAGTTGGAC-3′,引物序列由苏州金唯智公司合成。
1.3 PCR扩增引入酶切位点
测定第12位密码子MUT与WT质粒光密度(optical density,OD)值。依据OD值将MUT/WT 12按0∶1、1∶3 000、1∶10 000、1∶30 000的比例混合以模拟人体循环血液中游离DNA作为实验组模板,将MUT/WT 12质粒按0∶1、1∶300、1∶1 000、1∶3 000的比例混合作为对照组模板,WT质粒终质量浓度为0.02 μg/L[6]。以混合的人工模板进行PCR扩增,PCR反应体系为50 μL,5 μL 10×Tag buffer,0.4 mmol/L dNTP,引物FS、AS各20 mmol/L,Tag酶0.25 U,模板40 ng。PCR扩增条件:预变性95 ℃ 3 min;变性95 ℃ 22 s,退火58 ℃ 22 s,延伸72 ℃ 22 s,38个循环;终延伸72 ℃ 8 min。
1.4 限制性内切酶酶切
PCR产物纯化试剂盒纯化回收引入酶切位点的PCR产物,反应体系150 μL,PCR产物2 μg,12 μL 10×Cut Smart Buffer,5 μL BstNI,85 ℃反应2 h。PCR产物纯化试剂盒纯化回收酶切产物。野生型片段含酶切位点,被酶切成两个小片段,去磷酸化使5′端去磷酸根,与线性化载体的连接被阻断。突变型片段不含酶切位点,不能被酶切,去磷酸化处理不影响突变型片段与线性化载体的连接。因此,酶切后PCR产物去磷酸化。
1.5 酶切产物去磷酸化
纯化回收PCR酶切产物,反应体系60 μL,6 μL 10×碱性磷酸酶缓冲液,3 μL碱性磷酸酶,37 ℃ 1 h,80 ℃ 20 min以灭活碱性磷酸酶。PCR产物纯化试剂盒纯化回收去磷酸化产物。EcoRI与KpnI双酶切PMD-19T载体与PCR产物,纯化回收线性化载体与PCR酶切产物。紫外分光光度计测定OD值。
1.6 蓝白斑筛选检测稀有突变及蓝白斑菌落计数
测定PCR酶切产物的OD值,PCR酶切产物与载体连接。将连接后的产物导入E Coli.DH5α菌株。培养物涂抹于含X-gal、IPTG、Amp的LB琼脂平板培养基上,琼脂平板置于5%二氧化碳培养箱,37 ℃培养16 h。将培养皿平分为4个部分,肉眼计数1个部分的白色克隆与蓝色克隆,将所得计数的克隆数乘以4既为培养皿上的白色克隆或蓝色克隆的个数。
1.7 菌液PCR酶切鉴定插入片段
在突变与野生为1∶30 000琼脂培养皿上挑取5个白色克隆,进行菌液PCR酶切鉴定。引物AS与FS进行菌液PCR扩增以引入酶切位点,BstNI酶切,酶切产物琼脂糖凝胶电泳,将酶切鉴定为阳性的克隆一代测序,验证其插入片段的序列。
1.8 肺癌患者游离DNA验证
于南华大学附属第一医院采集26例肺癌患者外周血8 mL,血液置于EDTA抗凝管中。参照凯杰游离DNA提取试剂盒提取血中游离DNA[7]。利用限制性内切酶酶切联合去磷酸化与蓝白斑技术对26例肺癌患者游离DNA进行检测。
2 结 果
2.1 质粒引入酶切位点
BstNI酶切PCR产物,其电泳图见图1。野生型片段PCR扩增产物含CCWGG(W为A、C、T、G中任何一个碱基),为BstNI的酶切位点;突变型片段的PCR扩增产物不含CCWGG,不含酶切位点。PCR产物送金唯智公司测序,其序列图见图2。
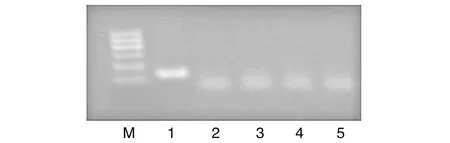
图1 质粒引入酶切位点酶切电泳图M为100 bp的Marker;1为PCR产物未酶切;2~5为MUT/WT 12分别为0∶1、1∶3 000、1∶10 000、1∶30 000酶切产物。
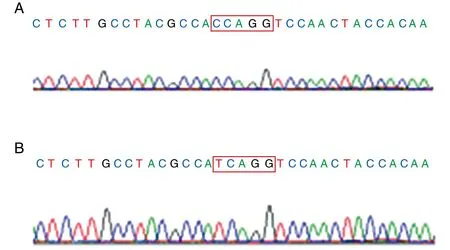
图2 人工模板引入酶切位点测序图A为野生型片段引入酶切位点;B为突变型片段引入酶切位点。
2.2 两组不同比例混合模板克隆结果
两组培养皿上均可见白色克隆与蓝色克隆。实验组酶切产物去磷酸化,被酶切成两个小片段的野生型片段与线性化载体的连接受到阻碍,突变片段为一完整片段,去磷酸化不影响突变片段与线性化载体的连接,与对照组相比较,白色/蓝色克隆大幅提升(图3)。
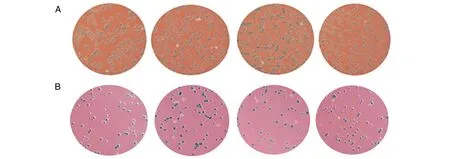
图3 不同比例混合模板的克隆形成图A从左到右依次为MUT/WT 12质粒比例分别为0∶1、1∶300、1∶1 000、1∶3 000克隆形成图;B从左至右依次为MUT/WT 12质粒比例分别为0∶1、1∶3 000、1∶10 000、1∶30 000克隆形成图。
2.3 蓝白斑菌落计数
对照组1∶3 000培养皿上白色克隆17个,蓝色克隆2 329个,白色/蓝色克隆为1/137;而实验组1∶30 000的培养皿上白色克隆23个,蓝色克隆394个,白色/蓝色克隆为1/17;1∶30 000实验组白色/蓝色克隆比例明显高于1∶3 000 对照组(表1)。

表1 蓝白斑菌落计数
2.4 菌液PCR酶切鉴定结果
实验组1∶30 000培养皿上的5个阳性克隆酶切鉴定出3个阳性克隆(图4A)。一代测序进一步验证插入片段的序列,其序列见图4B。野生型扩增片段为BstNI的酶切位点,被BstNI消化为80 bp与50 bp的两个DNA片段,这两个片段不能被琼脂糖电泳检测(可以用聚丙烯酰胺凝胶电泳检测)。

图4 菌液PCR酶切电泳图A为菌液PCR酶切电泳图(箭头所指为阳性克隆);B为阳性克隆插入片段序列图。a为80 bp片段;b为50 bp片段。
2.5 肺癌患者游离DNA验证结果
26例肺癌患者游离DNA检测出2例游离DNA为KRAS基因第12位密码子突变,其克隆形成图如图5A,测序验证其插入序列如图5B。

图5 肺癌患者游离DNA验证结果A为酶切联合去磷酸化与蓝白斑筛选对游离DNA检测的克隆形成图;B为酶切鉴定为阳性克隆插入片段的序列图。
3 讨 论
利用游离DNA进行肿瘤早期诊断与肿瘤切除术后复发监测时,富集低丰度突变DNA片段至关重要。微滴数字PCR检测热点突变能够在一万野生拷贝中检测出单一拷贝[8-9],但成本高,操作系统繁琐,现无法在基层医院推广为普通的体检项目。二代测序不能对肿瘤进行早期或超早期诊断,且测序费用高,不易被普通百姓接受[10-13]。Zhang等[14-15]发明的高保真聚合酶介导的引物3′末端硫化修饰技术虽检测下限可达到千分之一甚至更低,但具有假阳性并存的缺点。
蓝白斑筛选联合一代测序可检测千分之一的稀有突变,更适合于肿瘤切除术后复发监控[5]。对于一些体细胞突变,其野生型片段含酶切位点,突变引起酶切位点消失;或其野生型片段可经PCR扩增引入酶切位点,而突变型片段不能被引入酶切位点,可利用限制性内切酶酶切PCR扩增产物,去磷酸化,再与线性化载体连接,一代测序对阳性克隆的插入片段序列进行确定。野生型片段被酶切成两个小片段,去磷酸化处理,5′末端去磷酸基团的产物不能与线性化载体连接。对涉及终止密码子的突变,可通过克隆颜色对稀有突变进行初步区分。实验组MUT/WT 12为1∶30 000的培养皿上白色/蓝色克隆为1/17,而对照组MUT/WT 12为1∶3 000的培养皿上其白色/蓝色克隆为1/137,1∶30 000实验组白色/蓝色克隆比例明显高于1∶3 000对照组。野生型片段的PCR产物经限制性内切酶酶切,在T4连接酶的作用下,与线性化载体连接。蓝白斑筛选高效率与假阳性并存,对照组的培养皿上白色/蓝色克隆的比例高于实质模板比例,考虑为载体的自连。将实验组培养皿上的阳性克隆进行酶切鉴定,再将酶切鉴定为阳性克隆的一代测序以确定插入片段的序列可避免假阳性。本研究结果显示,限制性内切酶酶切后去磷酸化联合蓝白斑筛选可检测出1∶30 000 KRAS基因稀有突变,证明了限制性内切酶酶切后去磷酸化联合蓝白斑筛选可提高对稀有突变的检测能力。本实验结果表明限制性内切酶酶切后去磷酸化联合蓝白斑筛选在恶性肿瘤的早期或超早期诊断具有较大的应用潜力。
KRAS热点突变见于多种恶性肿瘤,其中,突变率最高的为胰腺癌,其次为直结肠癌、胆管癌及肺癌[7]。本课题组利用限制性内切酶酶切后去磷酸化联合蓝白斑筛选对KRAS基因第12密码子突变的人工模板进行检测,同时对26例肺癌患者游离DNA进行稀有突变检测以验证此技术的可行性。游离DNA检测为无创性检测,患者接受的意愿高。这一研究对涉及终止密码子的其他热点突变的检测具有潜在的临床应用价值,可为临床恶性肿瘤的诊断提供一种快速、准确的检测方法,对临床早期及超早期诊断肿瘤、确定治疗方案以及肿瘤的预后具有较重要的临床应用价值。此外,本方法操作简单,所需要的分子生物学试剂成本低廉,所需的仪器PCR仪、琼脂糖凝胶电泳等,均为分子生物学基本仪器,对操作人员的技术要求不高,有利于在基层医院普及。此项检测可纳入为常规的体检项目,为肿瘤的早期和超早期诊断提供一种低廉、低检测下限的检测方法,可以作为早期肿瘤筛查的检测技术。
