ICP-AES测量脱硝催化剂中五氧化二钒的不确定度分析
2024-02-23施宗友邵国庆
王 勇 施宗友 邵国庆 刘 林
(国家钒钛制品质量检验检测中心,攀西钒钛检验检测院,四川 攀枝花 617000)
脱硝催化剂主要用于烟气脱硝工艺,其原理是在脱硝催化剂作用下,以氨(或尿素)作为还原剂,将氮氧化物还原成氮气和水,从而达到脱硝的目的。目前研究和使用最多的是以二氧化钛为载体,以五氧化二钒为催化剂活性中心,并加入其它化学成分如氧化钨、氧化钼、二氧化硅、氧化铝等以提高催化剂综合性能[1]。催化剂中五氧化二钒含量是影响脱硝催化效率的关键之一,其含量大小将直接影响催化剂的性能[2,3]。目前针对脱硝催化剂中五氧化二钒的检测方法有X射线荧光光谱法(XRF)、电感耦合等离子体原子发射光谱法(ICP-AES)、磷钨酸分光光度法[4,5],这些方法均未对方法测量数据不确定度进行分析,在实际应用中无法判断测量数据的有效性,更不便于进行质量控制,有必要进一步讨论测量方法的不确定,为准确表达测量结果及质量控制提供依据。
不确定度作为测量结果定量评价的关键性参数,表征着测量结果的离散程序,与测量结果存在直接关联[6,7]。由于ICP-AES法具有测量线性范围广、多元素同时测量、分析速度快等优势,应用范围广[8-10]。本文在文献[4]方法的基础上,依据JJF 1059.1—2012《测量不确定度评定与表示》及分析检验过种中不确定度来源[11-13],结合ICP-AES法测定脱硝催化剂中五氧化二钒含量方法,分析测量过程不确定度来源及评定表达,最后,通过比较测量过程中各不确定度分量对合成不确定度的贡献大小,找到影响测量合成不确定度的关键来源,并提出有效控制措施,确保测定结果的准确性。对脱硝催化剂中五氧化二钒测量结果不确定的分析,不仅为其质量控制及测量结果准确表示提供依据,而且也为评定此类不确定度提供一种参考。
1 试验部分
1.1 试剂及材料
钒单元素标准储备溶液(国家标准物质研究中心):1 000 mg/L;盐酸(ρ约为1.19 g/mL):优级纯;硝酸(ρ约为1.42 g/mL):优级纯;氢氟酸(ρ约为1.15 g/mL):优级纯;二氧化钛:光谱纯;五氧化二钒:光谱纯;洒石酸:分析纯;酒石酸溶液:150 g/L;过氧化钠(分析纯)。
试验用水为超纯水。
1.2 仪器设备
Multiwave PRO型微波消解仪(安东帕(上海)商贸有限公司)。
Optima 8300型全谱直读等离子体发射光谱仪(美国PerkinElmer公司),配有耐氢氟酸系统。ICP-AES工作条件:高频发射功率为1 300 W;蠕动泵泵速为15 mL/min;等离子气体流量为15 L/min;雾化气流量为0.55 L/min;辅助气流为0.2 L/min;观测高度为15 mm。
移液器(德国普兰德):100~1 000 μL;电子天平(梅特勒-托利多公司),线性误差为±0.1 mg。
1.3 检测方法
1.3.1 样品处理方法
方案A:称取0.100 g(精确至0.000 1 g)试样于聚四氟乙烯消解罐,加入1 mL 150 g/L酒石酸溶液、2.0 mL 氢氟酸和3.0 mL硝酸,用少量水冲洗罐壁,盖紧消解罐,送入微波消解仪内,按设定的消解程序进行消解。试样消解完成后,取出冷却,将试液转移至100 mL容量瓶,以水定容,摇匀。
方案B:称取0.100 g(精确至0.000 1 g)试料置于刚玉坩埚,放入电阻炉内,由室温升至650 ℃,灼烧1 h,取出冷却后,加入3 g过氧化钠,用玻璃棒搅匀后,置于已升温至650 ℃的电阻炉内,待试样完全熔融后保持1~3 min,取出冷却。将坩埚放入250 mL烧杯,加入40 mL沸水置于电热板煮沸3 min后取下,将溶液转入至100 mL容量瓶定容后过滤,滤液待测。
1.3.2 标准曲线绘制及测定
按照方案A和B称取相应光谱纯二氧化钛,每份0.08 g(试样中二氧化钛按80%计算),按照1.3.1节的方案A和方案B进行消解后转移至100 mL容量瓶,用移液器加入钒标准储备溶液(1 000 mg/L)0、0.20、0.50、1.00、1.50、2.00 mL,以3%(V/V)硝酸定容,摇匀。此校准溶液系列(对应为A和B)中钒元素质量浓度为:0、2.0、5.0、10.0、15.0、20.0 mg/L。
2 数学模型
根据公式(1)计算试样中V2O5的含量。
(1)
式中:wx为试样中五氧化二钒的质量分数,%;ρ1为自校准曲线上查得试料溶液中钒的质量浓度,mg/L;ρ0为自校准曲线上查得空白溶液中钒的质量浓度,mg/L;V为测量溶液的体积,mL;m为试样质量,g;MV2O5为五氧化二钒摩尔质量,g/moL;MV为钒摩尔质量,g/moL。
3 不确定度来源识别
根据测定步骤和数学模型可知,脱硝催化剂中V2O5含量不确定度的主要来源包括试样称量、标准物质、校正溶液、校准曲线、样品溶液测量重复性、摩尔质量六个方面引入的不确定度,各不确定度分量计算讨论如下。
3.1 试样称量引入的不确定度Urel(m)
天平称量样品引起的标准不确定度分量urel(m)。称取0.100 g样品,天平的误差为0.1 mg,扩展不确定度为0.000 2 g,按均匀分布标准不确定度为0.000 12。称量样品0.100 g时,样品质量的相对标准不确定度为:
(2)
3.2 标准物质纯度的相对标准不确定度Urel(p)
查标准物质证书得到钒标准物质浓度p为1 000 mg/L,扩展不确定度u为1%(k=2)。标准物质的相对标准不确定度为:
(3)
3.3 标准溶液配制的相对标准不确定度Urel(std)
查阅JJG196—2006《常用玻璃量器检定规程》,20 ℃时100 mL容量瓶(A级)容量允许差为0.10 mL,查阅移液器(100~1 000 μL)检定证书,其容量允许差为0.005 mL,取均匀分布,得到由定值误差的标准不确定度u(Vval)。玻璃量器在25 ℃使用,与校准温度存在差异,水的膨胀系数2.1×10-4mL/℃,由温差引起的体积变化为ΔV=玻璃器具标称容量×溶剂膨胀系数×5,取均匀分布,得到由温度差异的标准不确定度u(Vtemp),玻璃量器的相对标准不确定度按公式(4)计算。
(4)
标准溶液配制过程中使用玻璃量器和移液器的不确定度分别为:
urel(Vflask10)=
(5)
urel(Vpipette)=
(6)
在配制过程中用到100 mL容量瓶6次,1 mL移液器5次,标准溶液配制的相对不确定度为:
(7)
3.4 工作曲线变动性的不确定度Urel(cal)
分别测定标准曲线A和标准曲线B,采用最小二乘法将钒的发光强度与对应浓度拟合标准曲线回归方程:I=aC+b,式中I代表标准曲线溶液中钒的发光强度,a为标准曲线斜率,b代表标准曲线截距,C为标准曲线溶液中钒的浓度。所得发光强度及相关参数见表1、表2。由线性回归方程求得试样中钒的质量浓度,标准曲线的标准不确定度为:
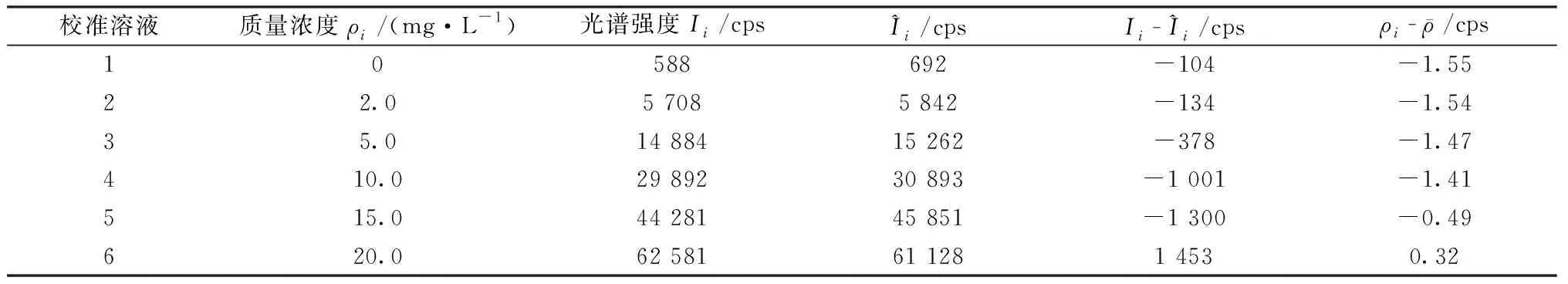
表1 A工作曲线和统计参数
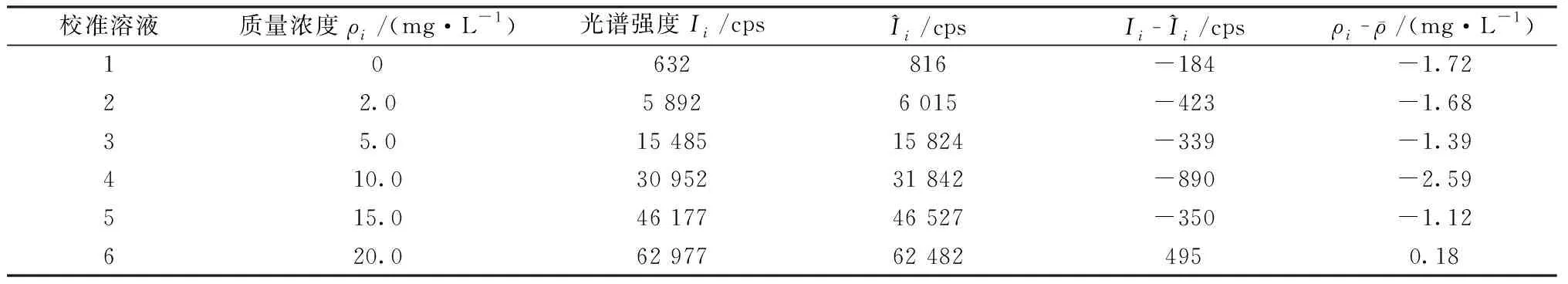
表2 B工作曲线和统计参数
(8)
(9)
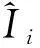
3.5 样品重复测量的相对标准不确定度Urel(frep)
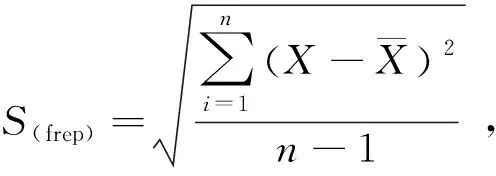
(10)

表3 试样溶液中钒的测量结果
由表3可知,方案A的测量结果分布(标准偏差)为0.040,小于方案B的0.101,因此方案A在试样前处理方面较好。
3.6 V2O5摩尔质量的相对不确定度Urel(M)

(11)
3.7 合成标准不确定度Urel(C)
根据上述分析,将电感耦合等离子发射光谱法测量脱硝催化剂中V2O5含量整个测量过程各不确定度分量见表4(各不确定度分量之和为100%)。从表4中各不确定度分量占比可以看出,标准溶液配制(约43%)、工作曲线(约24%)、样品重量测量(约23%)、称量(约10%),以上四个不确定来源是影响整个测量合成不确定度的关键环节。因为方案A和方案B所用标准物质、玻璃量具、称量天平相同,因此在A、B方案下,这几种不确定度可作相同处理。从两种不同消解方案对测量结果的影响来看,方案A工作曲线变动不确定度为0.002 3,方案B为0.003 7;从试样重复测量不确定度来看,A方案测量不确定度为0.001 43,B方案的为0.003 69,造成上述差异主要原因是方案A与方案B消解方式的差异,因为消解方法的差异,试样中待测元素转移到溶剂中也存在差异,比如试样是否消解完全、溶液中是否含有胶体,被测离子发生水解等,都会影响待测元素含量的测量值,从实验数据可以看出A方案优于B方案。测量结果的相对合成不确定度计算如公式(12):方案A合成不确定度为0.016,方案B合成不确定度为0.028。
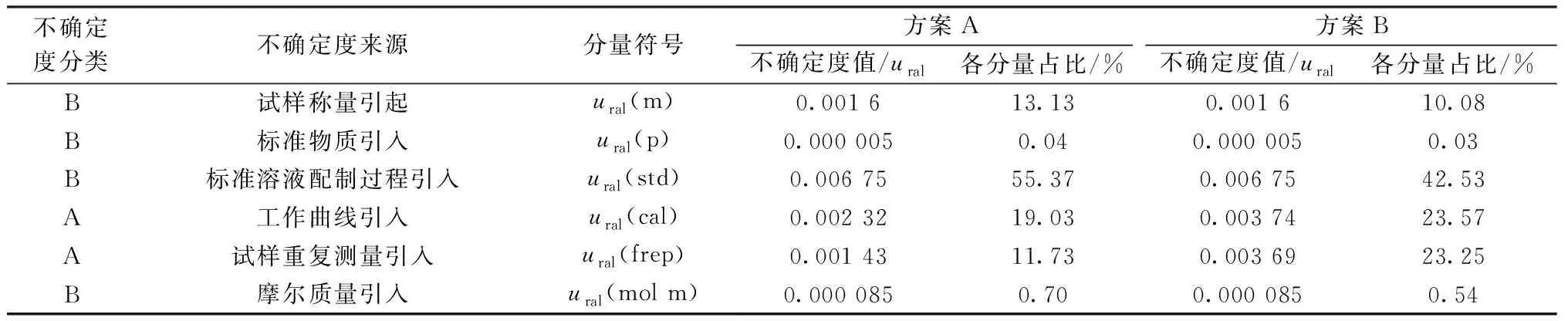
表4 各不确定度分量统计
(12)
由表4可知,在整个测量过程中,测量结果的最大不确定度来源为标准溶液配制过程、试样重复测量及标准曲线变动所引入的不确定度。
3.8 扩展不确定度(UA)及结果表示
取置信水平P=95%,包含因子K=2,则:A方案扩展不确定度uA=k×uc=0.016×2=0.032,B方案扩展不确定度uB=k×uc=0.028×2=0.056,两种方案试样中五氧化二钒的质量分数分别表述为方案A:ρ=(2.03±0.032)%;方案B:ρ=(1.99±0.056)%。
4 结论
测量过程中最大不确定度来源为标准溶液配制过程、试样重复测量及标准曲线变动引入的不确定度,在测量过程中应重点关注和控制这三方面引入的不确定度,具体措施建议:
1)尽量采用精度等级高的定量容器具,如A类容量瓶、移液管。
2)尽量减少试验过程稀释次数。
3)选用合适的样品消解方法,减少试样中等测元素的损失。
