HPLC法测定杨梅树不同药用部位的杨梅苷含量
2024-02-22邹福贤范世明
刘 娥,邹福贤,范世明
(1.福建医科大学附属第二医院,福建 泉州 362000;2.福建中医药大学范世明中医药传承工作室,福建 福州 350122;3.福建中医药大学附属泉州市正骨医院,福建 泉州 362000;4.福建中医药大学药学院,福建 福州 350122)
杨梅(Morella rubra Lour.)的栽培历史超过2 000 年,药用历史超过1 700 年,杨梅根、根皮、树皮、枝、叶和果实均可入药[1]。《本草纲目》记载杨梅果实去痰、止呕哕、消食下酒,树皮及根主治恶疮疥癞、牙痛。此外《贵州草药》《江西民间草药验方》《福建药物志》也均有记载杨梅不同药用部位的功效主治,主要为行气活血、通关开窍、消肿解毒,主治跌打损伤、骨折、外伤出血等疾病[2-4]。杨梅主要含有丰富的黄酮类成分,如杨梅苷、杨梅素、杨梅醇、槲皮素、槲皮苷、芦丁和山柰素等,其中杨梅苷含量远高于其他成分[5-8]。现代药理研究表明杨梅苷具有抗菌[9]、抗氧化[5,10]、抗病毒[11]、抗肿瘤[12]、降血糖[13]及镇痛[14]作用,能够抑制破骨细胞分化[15],改善心肌收缩功能,抑制细胞凋亡[16],说明杨梅苷为杨梅树的活性成分之一。
然而,目前关于杨梅的研究多集中在其果实风味,关于其不同药用部位活性成分的研究较少[17]。此外杨梅树每年均需修枝剪叶,产生大量的杨梅树药材资源,若能充分利用起来将有利于中药资源的可持续发展,避免极大的浪费,因此本研究拟对杨梅树的不同药用部位进行杨梅苷含量测定,为深入开发杨梅药用资源奠定基础。
1 材 料
1.1 仪器 KQ-500VDY 型医用超声波清洗器(昆山超声仪器有限公司);PX225DZH 十万分之一电子分析天平[奥豪斯仪器(上海)有限公司];FA1204B 万分之一电子分析天平(上海菁海仪器有限公司);Essentia LC-16 高效液相色谱仪(日本SHIMADZU 公司;SPD 紫外检测器、Milli-Q 超纯水仪均购自德国Merck 集团。
1.2 试剂与药材 杨梅苷对照品(中国食品药品检定研究院,纯度≥95.2%,批号:111806-201703);色谱纯乙腈(天津市康科德科技有限公司);色谱纯甲醇(国药集团化学试剂有限公司);超纯水经Milli-Q 超纯水仪处理;3 批杨梅树样品均采自福建省南安市,经范世明正高级实验师鉴定其基源为杨梅科植物杨梅(Morella rubra Lour.)。杨梅果根据其颜色、硬度和大小分为5 个成熟阶段:杨梅果Ⅰ期表面呈现淡粉色和青色,缝合线明显,肉柱连接紧密,果实直径<1 cm;杨梅果Ⅱ期表面淡粉色,略有泛白,肉柱连接紧密,果实直径与Ⅰ期变化不大;杨梅果Ⅲ期表面粉色,部分红色,肉柱连接紧密,果实开始膨大;杨梅果Ⅳ期表面红色,色泽均匀,肉柱软硬适中,果实直径超过1 cm;杨梅果Ⅴ期表面为暗红色,色泽均匀,肉柱软润饱满,果实直径超过1 cm[18-20]。见图1。

图1 杨梅果的不同分期
2 方法与结果
2.1 供试品溶液制备 分别取3 批杨梅树的不同药用部位(根木质部、根韧皮部、树韧皮部、枝韧皮部、枝木质部、叶、果)的干燥粗粉各3 份,每份0.5 g,精密称定后置于锥形瓶中,加入甲醇30 mL;称定整个锥形瓶及溶液质量,随后置于超声波清洗器中超声(40 ℃,45 kHz)处理30 min;取出锥形瓶,冷却至室温,再称定整个锥形瓶及溶液质量;用甲醇补足减失的质量,摇匀,将溶液过0.45 μm 微孔滤膜,取过滤的中段溶液作为供试品溶液。
2.2 对照品溶液制备 精密称取纯度为95.2%的杨梅苷对照品10.22 mg 置于10 mL 量瓶中,加入甲醇溶解,定容,摇匀,制成0.972 9 mg/mL 溶液,作为对照品溶液。
2.3 色谱条件 色谱柱:Ultimate®XB-C18(4.6 mm×250 mm,5μm);乙腈-0.1%磷酸水溶液(20∶80)等度洗脱10 min,设置流速为1 mL/min,柱温为40 ℃,检测波长为260 nm,进样量为10 μL。分别取不同药用部位的供试品溶液和对照品溶液,按上述色谱条件进样,色谱图见图2。结果显示:杨梅苷色谱峰对称,与相邻色谱峰分离度良好,基线平稳,理论板数均不低于20 000,表明该方法专属性强。

图2 杨梅不同药用部位样品色谱图
2.4 线性关系考察 精密吸取“2.2”项下配制的对照品溶液适量,加甲醇逐级稀释,摇匀,分别配成3.891 8、11.675 0、19.459 0、48.647 0、97.294 0、194.590 0 μg/mL 对照品溶液,按“2.3”项下色谱条件测定,测定结果以杨梅苷浓度(X)为横坐标,以峰面积(Y)为纵坐标,绘制标准曲线,计算得到线性回归方程为Y=2.787 6×104X-2.461 0×104,r=0.999 9,表明杨梅苷浓度在3.891 8~194.590 0 μg/mL内与峰面积呈良好线性关系。
2.5 精密度考察 精密吸取“2.4”项下配制的48.647 0 μg/mL 对照品溶液,按“2.3”项下色谱条件连续测定6 次,记录峰面积。结果显示:杨梅苷的峰面积RSD 为0.17%,表明仪器精密度良好。
2.6 稳定性考察 通过对杨梅根的韧皮部和木质部进行质量检测,发现杨梅根中韧皮部与木质部的质量占比为15∶85,故将根韧皮部与根木质部粗粉按质量比15∶85 混合均匀作为混合样品。取1 份混合样品,按“2.1”项下方法制备供试品溶液,分别于室温下放置0、2、4、8、12、24 h,按“2.3”项下色谱条件进行测定,记录峰面积。结果显示:杨梅苷的峰面积RSD 为0.260 0%,表明供试品溶液在室温下放置24 h 内稳定性良好。
2.7 重复性考察 取“2.6”项下方法制作的混合样品6 份,每份0.500 0 g,按“2.1”及“2.3”项下方法制备供试品溶液并进行含量测定,记录峰面积,按线性回归方程计算样品中杨梅苷含量。结果显示:6份样品杨梅苷平均含量为0.240 0%,RSD 为1.590 0%,表明本方法重复性良好。
2.8 准确度考察 取“2.6”项下方法制作的混合样品6 份,每份0.250 0 g,精密称定,再分别向每份样品中精密加入浓度为0.972 9 mg/mL 杨梅苷对照品溶液0.600 0 mL。按“2.7”项下杨梅苷平均含量占比结果计算样品中杨梅苷原有含量,按“2.1”及“2.3”项下方法制备供试品溶液并检测杨梅苷含量,同时计算回收率。结果显示:杨梅苷的平均回收率为97.703 5%,RSD 为2.271 3%,表明本方法准确度良好。见表1。
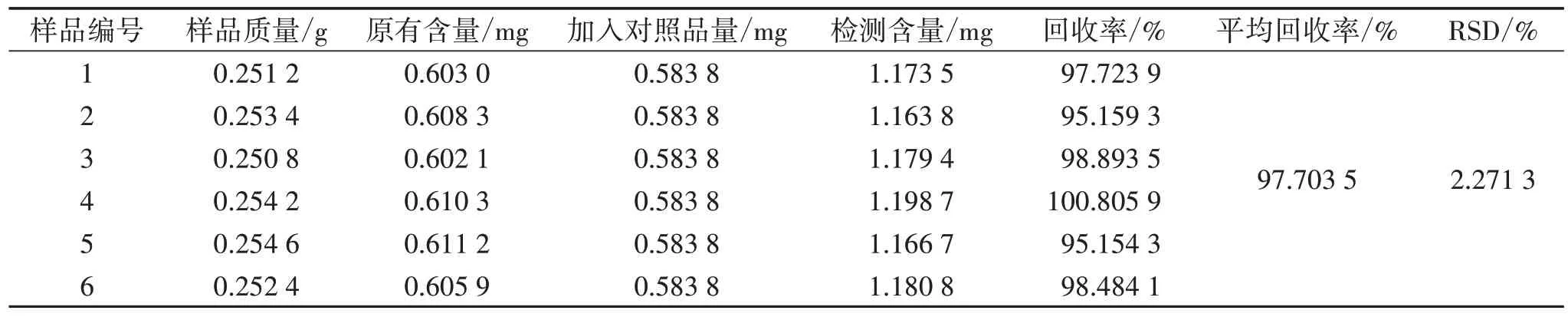
表1 杨梅根部杨梅苷含量测定方法准确度考察
2.9 含量测定 分别取3 批杨梅的不同药用部位样品各3 份,按“2.1”及“2.3”项下方法制备供试品溶液并进行含量测定,记录峰面积,按线性回归方程计算样品中杨梅苷含量,记录3 批杨梅不同药用部位样品的平均值,见表2。结果显示:杨梅不同药用部位中杨梅苷含量在0.043 0%~13.917 0%,各个药用部位杨梅苷含量差别较大,树韧皮部>根韧皮部>枝韧皮部>叶>枝木质部≈根木质部>果;杨梅果随着成熟度的增加,杨梅苷含量在逐渐降低。
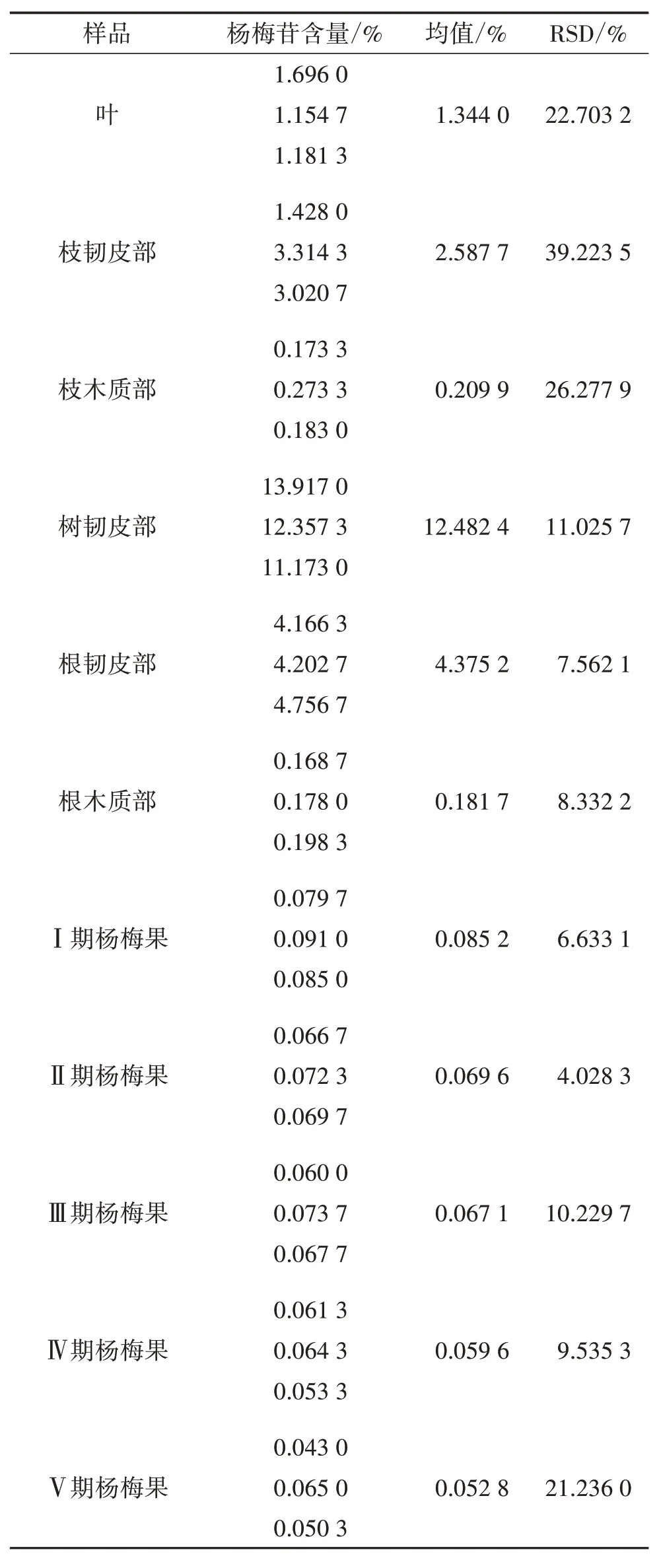
表2 杨梅树不同药用部位中杨梅苷含量测定结果
3 讨 论
3.1 HPLC 方法优化 不同文献中杨梅苷含量测定时所选择的检测波长并不相同,有260、358、360 nm[21-24]。因此,为确定杨梅根中杨梅苷的最佳检测波长,本研究将供试品溶液和对照品溶液在200~400 nm 波长范围内进行检测,发现杨梅苷在225、260、350 nm 处有波峰,而225 nm 处于紫外检测末端,容易产生干扰,260 nm 处吸收强度高于350 nm,且供试品溶液在260 nm 下杨梅苷色谱峰附近无其他色谱峰干扰,因此选择260 nm 作为检测波长,检测结果更加灵敏。此外,本研究所确定的流动相洗脱程序为等度洗脱,相比于其他文献中的梯度洗脱[20],基线更为平稳,检测结果更加准确。
3.2 杨梅不同药用部位杨梅苷含量比较 有文献报道杨梅叶中杨梅苷含量为1.430 0%[21],果实中含量为0.041 3%[22],与本研究所测得结果较为接近。通过对杨梅不同药用部位的杨梅苷含量测定发现:杨梅苷主要集中在韧皮部,而又以树韧皮部中含量最高,是根韧皮部或者枝韧皮部的3~5 倍;果实中含量最低,平均含量在0.066 9%,仅为树韧皮部的1/200。因此,根据杨梅苷的药理作用,若要发挥其抗菌、降血糖、镇痛等方面的疗效,应当选择杨梅树韧皮部入药。
综上所述,本研究建立了一种简便快捷的HPLC 方法以便于测定杨梅树中杨梅苷含量,该方法明确了杨梅树不同药用部位中杨梅苷含量的分布,其中以韧皮部和叶中含量最高,这为杨梅果树修枝剪叶所产生的大量枝条再利用和深入开发提供了实验基础,有助于中药资源的可持续发展。
