国内基因编辑疗法获批还有多远?
2024-02-18周游
周游

本文图/视觉中国
北京时间2023年12月9日,美国食品药品监督管理局(FDA)宣布批准一款商品名为Casgevy的CRISPR/Cas9基因编辑疗法上市,用于治疗12岁及以上的镰刀型细胞贫血病(SCD)患者,定价220万美元,折合人民币超过1500万元。此前2023年11月17日,英国药品和健康产品管理局已批准该疗法上市,Casgevy也是全球首个获批的基因编辑疗法。理论上,CRISPR/Cas9基因编辑体系能让科学家精准地对人类DNA进行改造,一劳永逸地抵御基因性疾病。
相比Casgevy在国外的火热,清华大学药学院创始院长、全球健康药物研发中心主任丁胜向《中国新闻周刊》分析,目前国内非上市药企是入局基因编辑疗法的主力,而上市药企观望居多。国内基因编辑疗法的研发尚处于临床早期,离获批上市还有一段距离。
美国FDA2023年6月宣布接受Casgevy的生物制品许可申请,到2023年12月获批仅用时6个月。根据1996年修订的美国联邦《食品、药品、化妆品法案》,新药上市的标准评审时间为10个月,可见FDA对其启用了优先、快速审评。上海科技大学生命科学与技术学院教授、基因编辑中心主任陈佳告诉《中国新闻周刊》,这与Casgevy的核心技术体系CRISPR/Cas9的革命性是分不开的。
陈佳表示,CRISPR/Cas9是当前最主流的基因编辑体系。该技术又称“基因剪刀”,于2012年由法国科学家埃马纽尔·夏彭蒂耶和美国科学家詹妮弗·杜德纳在《科学》杂志上首次提出。2020年,二人因CRISPR/Cas9的广阔应用潜力,共同获得当年的诺贝尔化学奖。此次Casgevy的许可申请由美国福泰制药与瑞士基因编辑公司CRISPR Therapeutics联合提出,后者的创始人正是夏彭蒂耶。
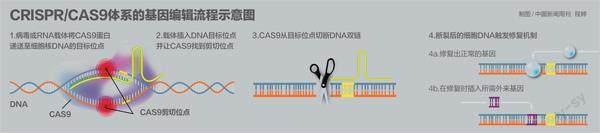
CRISPR/Cas9体系存在于原核生物的细胞中,原核生物主要指细菌。陈佳介绍,CRISPR/Cas9体系在体外能定点对DNA进行剪切,在活细胞中有修改基因的能力。CRISPR是细菌DNA里特殊的一段。某些细菌遭到病毒入侵后,能够把病毒基因的一小段存储为CRISPR。病毒再次入侵时,细菌能根据这段“记忆”识别病毒,用Cas蛋白将病毒的DNA切断。Cas蛋白是一种核酸酶,能够对目标基因位点进行剪切编辑,相当于“剪刀”。目前最常用的Cas蛋白为Cas9。
FDA官网显示,Casgevy的适应证包括SCD和β-地中海贫血症(TDT)。其在英国获批时申报的适应证包括SCD和TDT两项,而此次FDA审批的适应证仅有SCD,针对TDT的上市获批时间预计为今年3月。美国伦斯勒理工学院化学生物学博士、正序(上海)生物科技有限公司CEO牟晓盾向《中国新闻周刊》介绍,SCD和TDT的病因类似,都是遗传性血红蛋白病。其中SCD患者的血红蛋白变得容易粘连,使得红细胞从正常的扁圆形转变成镰刀状。这些镰状红细胞僵硬、变形性差,会堵塞血管造成疼痛,严重时产生血栓,威胁患者生命。SCD主要发病群体在非洲。TDT则是由于血红蛋白合成障碍导致的溶血性贫血,患者极度依赖输血。
北京师范大学中国公益研究院2021年发布的《中国地中海贫血蓝皮书》显示,TDT在国内南方省份发病率较高,是出生缺陷防治重点疾病。2020年,全球TDT基因携带者约3.45亿人,中国携带者约3000万人,中、重度患者人数总计约30万,并正以每年约10%的速度递增。2023年11月,国家卫健委印发通知,在广东、广西等10个省份组建全国TDT防控协作网,继续推进TDT的免费产前筛查,降低重症TDT患儿的出生概率。
牟晓盾称,传统的TDT治疗手段为输血,患者长期依赖输血,可能会面临血源紧张和中断等问题。想要根治则需诉诸造血干细胞移植,也就是将具有正常血红蛋白基因功能的干细胞移植到患者体内,以补偿血红蛋白的表达,理论上可实现永久性治疗。但移植疗法依赖血液配型,存在配型成功率低、等待时间长、年龄限制等问题。如今,Casgevy的获批提供了全新的治疗策略,能帮助患者摆脱输血依赖,实现“一次治疗,终身治愈”。
CRISPR/Cas9体系究竟在治疗中发挥了什么作用?丁胜介绍,胎儿血红蛋白是人体内健康的、可以正常携带氧气的血红蛋白,但其只会在胎儿发育过程中产生,胎儿出生后,它的表达通道就被关闭了。研究显示,该表达通道被人类染色体上的BCL11A基因所抑制,当这一基因突变失去活性后,抑制失效,通道打开,人体又能制造胎儿血红蛋白了。对健康人群来说,出生前供氧依靠的是胎儿血红蛋白,出生后则依靠成人血红蛋白。SCD和TDT患者的成人血红蛋白有基因缺陷,直接编辑相应基因又难以实现。因此,可以借助CRISPR/Cas9对患者造血干细胞的BCL11A基因进行切割,模拟上述突变,相当于“松开了胎儿血红蛋白的刹车”。
2021年,《新英格兰医学杂志》发文展示了第一批接受CRISPR/Cas9體系治疗患者的结果。其中一位患者在接受治疗6个月内,胎儿血红蛋白在红细胞中的比例从治疗前的10.1%升至99.7%,并且能维持18个月。另一位患者的这一比例则在5个月升至99.9%,到第15个月还能维持近100%。这意味着在长期随访中,患者体内能检测到高水平的胎儿血红蛋白,不再需要接受输血治疗。Casgevy获批当日,福泰制药公布了一项针对SCD的临床试验,29名接受了Casgevy治疗的长期随访患者中,有28人在治疗后至少一年内没有SCD相关的严重疼痛症状出现。在当日福泰制药公布的另一项针对TDT的临床研究中,42例长期随访患者中有39例在治疗后至少一年内不需要输血,其余3人的输血需求减少了70%以上。
2023年3月召开的第三届人类基因组编辑国际峰会上,首位接受Casgevy治疗的SCD患者维多利亚·格雷发表了讲话。她称自己在接受治疗前,需要不时到医院接受输血、使用强效止痛药等,难以维持正常工作和生活。接受治疗近一年后,她没有做过输血治疗,疼痛几乎消失。她2023年的骨髓细胞活检显示,超过80%的造血干细胞产生了预期的遗传变化,证明了基因编辑治疗的长期有效性。
在陈佳看来,基因编辑治疗区别于普通基因治疗,后者是一种替代疗法。替代疗法的思路是,针对异常突变的基因,使用病毒载体向细胞里递送一段没有突变的基因,代替原本的基因发挥功能,也就是“缺啥补啥”。基因治疗中常用的病毒载体是腺相关病毒(AAV),目前FDA批准的基于AAV的基因疗法已有两项,分别用于治疗遗传性视网膜疾病和脊髓性肌萎缩症。
牟晓盾指出,替代疗法有三个明显的缺陷。一是病毒递送进去的“替补基因”编码蛋白的能力不是永久的,失效时间长则两三年,短则半年;二是病毒递送的方法和病毒感染细胞相似,都是随机插入细胞的DNA中,已被证实会产生副作用;三是人体会对病毒载体产生抗药性,失效后的二次治疗效果通常不理想,因此替代疗法既不能终身治愈,也不能重复治疗。
而基因编辑是在人体细胞DNA上进行修改而不是替代。牟晓盾解释,基因编辑疗法递送给细胞的不是外部合成的基因,也不一定需要病毒載体,而是CRISPR/Cas9这样的基因编辑器。CRISPR/Cas9可以进入细胞内部对目标基因定点编辑,放弃了短寿的“替补基因”。编辑好的基因永远存留在细胞内,因此,基因编辑可以做到终身治愈。此外,Cas9蛋白能在48~72小时内于细胞中自然降解,不会像插入DNA中的病毒一样留下安全隐患。
但CRISPR/Cas9体系的安全性同样值得关注。陈佳指出,Cas9作为“基因剪刀”,每一次编辑都会造成细胞DNA双链的断裂。DNA断裂将触发细胞的凋亡机制,细胞可能出现大量死亡。
“这本来是好事。”牟晓盾解释,细胞凋亡其实是对人体的保护,是清除遗传物质异常的细胞的一种机制。但同时,经过Cas9剪切还能存活下来的细胞,很有可能是凋亡机制出问题的细胞,这些“不死”的细胞将面临着演化成癌细胞的风险。因此,CRISPR/Cas9相当于对人体细胞进行了一次逆向筛选,把危险细胞留下来了。陈佳称这一过程为“劣币驱逐良币”。
牟晓盾指出,虽然FDA在审批文件中提到不要让完美主义来阻挡科技的发展,但必须承认CRISPR/Cas9体系可能对患者的长期健康造成负面影响。因此,目前所有的基因类药物和疗法,不管是基因治疗还是基因编辑治疗,FDA都建议要观察随访15年以上。基于CRISPR/Cas9体系的基因编辑疗法的安全性还有待更长期的随访研究。
2023年9月,《新英格兰医学杂志》报道了一位杜氏肌营养不良症患者在接受CRISPR/Cas9体内基因编辑治疗后死亡的案例,研究显示死因是该患者对高剂量的AAV载体产生了严重的免疫反应。
牟晓盾指出,基因编辑分为体外编辑和体内编辑两种,前者是指取出患者细胞在体外进行编辑后再回输体内,Casgevy即为体外编辑疗法,后者是指通过注射等方法递送基因编辑系统至目标器官进行编辑。目前,体内编辑的主要递送载体还是病毒,因此,CRISPR/Cas9体系还无法完全摆脱病毒递送的副作用。
另一影响安全性的因素是CRISPR/Cas9体系的脱靶率。FDA官网显示,在Casgevy获批上市前,FDA 曾召集专家委员会讨论 Casgevy 潜在的脱靶问题,最终专家组以全票支持,认为两家申请公司已做出了充足的脱靶效应分析,但依然建议对接受治疗的患者执行长期随访。
牟晓盾将脱靶比喻为“导航失误”。在普通基因治疗中,基因递送的准确性主要依赖于病毒载体,基因是车,病毒就是导航。很多时候导航不够精确,可能将基因送去了别的器官,也即脱靶。陈佳进一步指出,基因编辑中的脱靶存在两个层面,一是上述递送工具的脱靶。递送脱靶可能将本应递送到肝的编辑器递送到了脾,一般不会造成安全性问题,只会影响编辑效率,使得针对肝脏的疗效会下降。
另一个层面是编辑器脱靶。陈佳称,编辑器脱靶更像是编辑器自己“业务不精”,在细胞内编辑目标基因时,同时编辑了非目标基因。假设目标器官为肝,编辑器因递送脱靶到脾后,如果自身还存在编辑脱靶,很可能引发难以预料的健康问题。牟晓盾指出,CRISPR/Cas9体系目前依然存在编辑脱靶问题。对于一个基因编辑位点,它在整个细胞基因组中可能存在上千个相似的位点,相似度达到75%以上就会被同时编辑。这也为该技术的安全性埋下了隐患。
虽然CRISPR/Cas9体系能做到终身治愈,其治愈过程却常常令患者感到痛苦。陈佳介绍说,在确定病因例如TDT后,患者首先要进行造血干细胞的动员。造血干细胞平时只住在脊髓里,在注射动员剂后,一部分造血干细胞从骨髓进入血液循环,就可以通过静脉采血获得造血干细胞样本,进行体外编辑。患者在接受编辑好的造血干细胞移植前还需进行清髓,即通过药物将骨髓里残留的造血干细胞杀死,为健康细胞腾出位置。
由于清髓需要使用化疗药物,患者在此阶段会经历一定副作用,例如免疫力降低、器官疼痛等。陈佳称,这种疼痛有时非常严重,老年患者或已有器官受损的患者可能无法忍受。另外考虑到前述细胞凋亡机制,CRISPR/Cas9体系要求采集的干细胞数量非常多,才能达到一定数量的有效编辑,有些患者的身体状况可能负担不起。
镰状细胞贫血症血管三维示意图。
在陈佳看来,要规避体外编辑在治疗中对患者造成的痛苦,就必须在体内编辑的递送方面下功夫。如果体内编辑能有革命性突破,基因编辑能应用到更多的疾病治疗。体内编辑的技术国内外均有研发,目前,这类递送主要集中在肝脏,肝外器官的精准体内编辑还较难实现。
除了递送方式,编辑器的设计和迭代也是基因编辑疗法未来努力的方向。陈佳指出,目前所有编辑器都没能实现长片段的DNA的原位替换或修正。一般来说,500至1000个碱基以上就属于长片段,而当前很少有能编辑长度超过100个碱基片段的编辑器。碱基是DNA的基本组成单位。如果能做到精准递送、长片段递送,将极大拓展基因编辑疗法的适应证范围。
丁胜表示,国际上的基因编辑疗法除了Casgevy外,其他多处在临床早期阶段,离获批也许还有数年时间。他认为,Casgevy仅把CRISPR/Cas体系用于体外编辑,本质上还是一种细胞疗法,其编辑方法成熟,容易实现,但无法精确到单独的碱基编辑。DNA双链切断后的自动修复过程也不完全可控,可能存在双链接错等一系列问题。精准基因编辑体系,特别是体内编辑体系还处于早期研发阶段,离上市还有很长距离。
公开资料显示,早在2003年,全球首款基因治疗药物“今又生”在中国获批上市,用于增强多种抗癌基因活性,由深圳市赛百诺基因技术有限公司开发。直到2017年美国FDA才批准第一款基因疗法Luxturna。2021年,国家药监局批准了国内首个血友病AAV基因治疗药物进入临床试验。仅2023年,截至11月,国内就有共30款AAV递送的基因治疗药物的临床试验获批,其中3款进入III期临床。
在基因编辑领域,国内虽然已有50余家公司涉足,但其中依然以初创公司为主,还未登陆资本市场,研发也多在临床早期,如博雅辑因、邦耀生物、本导基因等。例如博雅辑因生物科技有限公司研发了基于CRISPR/Cas9体系的TDT基因编辑疗法ET-01,是中国首个获国家药监局批准开展临床试验的基因编辑疗法,已于2022年11月完成I期临床试验。其余多家非上市药企都有基因编辑疗法在临床早期阶段酝酿。与此同时,上市药企在该领域却集体缺席。
在陈佳看来,这并不意味着国内技术水平的落后,甚至在一些细分领域国内还能反超国外。2015年,中山大学生命科学学院黄军就团队发表了全球第一篇使用CRISPR/Cas9技术对TDT治疗成果的文章。2023年5月,陈佳团队在国际期刊《自然-细胞生物学》上发表了其构建的新型碱基编辑系统,可以实现单个碱基的精准编辑。今年11月,正序生物与合作研究团队在《细胞干细胞》杂志发表了一种基于变形式碱基编辑器(tBE)的TDT治疗新策略。tBE已通过国际专利申请统一程序,在中、美、澳大利亚等15个国家和地区获得专利授权,这在国内自研碱基编辑工具中尚属首次。
牟晓盾指出,目前国内在研的基因编辑疗法基本上都是Casgevy的迭代,核心工具都是CRISPR/Cas9。有的产品可能在基因编辑位点上做出了优化,但CRISPR/Cas9本身的技术短板很难解决。tBE体系的优势在于其完全消除了编辑脱靶,摒弃了病毒递送,单碱基编辑也避免了“基因剪刀”造成的DNA双链断裂,整体安全性优于CRISPR/Cas9。牟晓盾透露,该疗法的上述优势已在细胞、器官、动物和人体内得到验证,今年1月将向国家药监局申请开展I期临床试验。
丁胜表示,目前,国内的监管审评体系已与国际标准接轨。在国家药监局、卫健委和科技部等多部门的监管体制下,人体干细胞、基因诊断与治疗等技术开发和使用的风险完全可控。牟晓盾称,安全合规永远是企业应守住的底线,企业希望跟审评机构的专家一起,按照符合国家和全球的制药标准的方式推进审批进程。但相比FDA,国内审评资源偏少,专家工作繁重。
相比监管,伦理审查则更为严格。2018年,南方科技大学生物系原副教授贺建奎因“基因编辑婴儿”事件,招致国内外广泛批评,贺建奎也在2019年因非法行医罪获刑三年。《中国新闻周刊》了解到,这一事件后,国内基因编辑相关研究的临床试验被大规模叫停,两年后才陆续重启。
丁胜提醒,目前国内外关于基因编辑的伦理边界划分是一致的,任何可遗传的人类基因编辑都不允许进入临床应用。贺建奎事件涉及胚胎的编辑,显然触犯红线。但Casgevy只涉及造血干细胞的编辑,其编辑后的性状是不会遗传。牟晓盾称,药企在与医院进行临床试验合作时,医院会有伦理委员会对试验的伦理风险进行审核。总体来说,伦理审查和监管审评一样,有据可依,只要不触犯红线,还是比较顺利的。
陈佳指出,上市药企缺席的主要原因依然是风险和收益的老问题。在前沿科技领域,研发属于高风险行为,这导致国内上市药企对基因编辑领域保持观望态度。丁胜表示,跟普通药物相比,个体化的基因编辑治疗前期研发门槛高,基因的设计、优化和载体选择都存在试错过程,每一个基因编辑后的细胞药物都需要质检,才能保证安全。这就导致此类药品难以批量化生产,成本居高不下。天价基因编辑疗法的背后是极其烧钱的创新过程和极高的沉没成本,这对于依赖融资的非上市药企来说存在不小风险。
