Interplay between microglia and environmental risk factors in Alzheimer’s disease
2024-02-11MiaopingZhangChunmeiLiangXiongjinChenYujieCaiLiliCui
Miaoping Zhang, Chunmei Liang, Xiongjin Chen, Yujie Cai, Lili Cui
Abstract Alzheimer’s disease, among the most common neurodegenerative disorders, is characterized by progressive cognitive impairment.At present, the Alzheimer’s disease main risk remains genetic risks, but major environmental factors are increasingly shown to impact Alzheimer’s disease development and progression.Microglia, the most important brain immune cells, play a central role in Alzheimer’s disease pathogenesis and are considered environmental and lifestyle “sensors.”Factors like environmental pollution and modern lifestyles (e.g., chronic stress, poor dietary habits,sleep, and circadian rhythm disorders) can cause neuroinflammatory responses that lead to cognitive impairment via microglial functioning and phenotypic regulation.However, the specific mechanisms underlying interactions among these factors and microglia in Alzheimer’s disease are unclear.Herein, we: discuss the biological effects of air pollution, chronic stress, gut microbiota, sleep patterns, physical exercise, cigarette smoking, and caffeine consumption on microglia; consider how unhealthy lifestyle factors influence individual susceptibility to Alzheimer’s disease; and present the neuroprotective effects of a healthy lifestyle.Toward intervening and controlling these environmental risk factors at an early Alzheimer’s disease stage, understanding the role of microglia in Alzheimer’s disease development, and targeting strategies to target microglia, could be essential to future Alzheimer’s disease treatments.
Key Words: Alzheimer’s disease; chronic stress; environmental factor; gut microbiota; microglia;particulate matter with diameter < 2.5 µm
Introduction
Alzheimer’s disease (AD), the most common cause of dementia, is a neurodegenerative disease characterized by memory loss and progressive cognitive impairment (Leng and Edison, 2021).Other clinical AD symptoms include language dysfunction, visuospatial difficulties, and personality changes, which seriously reduce patient quality of life and cause heavy social burdens (Holtzman et al., 2011).According to Alzheimer’s Disease International, globally, 75% of patients with dementia are undiagnosed, and in some low- and middle-income countries, this proportion may be as high as 90%.With their latest data, the World Health Organization estimated that in 2019, 55 million people suffered from dementia; this number is expected to rise to 139 million by 2050 (No authors listed, 2023).Primary AD pathology includes brain amyloid plaque depositions, which are formed by amyloid-β (Aβ) aggregation and neurofibrillary tangles triggered by tau hyperphosphorylation (Villemagne et al., 2013; Rickman et al., 2022; Roda et al., 2022).
Microglia, brain myeloid cells, act as the first-line immune protection in brain injury and disease (Estudillo et al., 2023).In the 1990s, Caudros et al.first suggested that microglia originate from the yolk sac during primitive hematopoiesis and fill the central nervous system (CNS) before vasculogenesis(Cuadros et al., 1993).From a morphological perspective, microglia are classified as ramified (resting), activated, or ameboid (phagocytotic).Increasing evidence indicates that microglia present a diverse range of phenotype states during chronic inflammation, which are categorized as:proinflammatory M1 and activated M2 (Orihuela et al., 2016; Ransohoff,2016).However, the M1/M2 dichotomy is criticized as an oversimplification that cannot modelin vivoconditions (Hansen et al., 2018).Current investigators increasingly stress the importance of studying various microglial phenotypes due to the complex process of microglial activation, and have revealed several new microglial phenotypes associated with AD.These include dark microglia, which are abundant in AD pathology, chronic unpredictable stress, and ageing (Bisht et al., 2016); disease-associated microglia (DAM),which re observed in AD and localized near Aβ plaques (Keren-Shaul et al.,2017); microglial neurodegenerative phenotype, which are isolated from the brain and spinal cord of amyloid precursor protein (APP)-PS1 mice (Bisht et al., 2018); activated response microglia, which are enriched with AD risk genes (Sala Frigerio et al., 2019); and human Alzheimer’s microglia (Srinivasan et al., 2020).Clearly, microglia are likely key participants in AD.
AD is a chronic disease influenced by both the external environment and genetics.The latter play an important role in AD development (Silva et al.,2019).Gene mutations such as APP or PS1 can directly lead to familial AD.There is consensus that the ε4 allele of the apolipoprotein E gene (APOE4) is the most significant genetic risk factor for sporadic AD (Martens et al., 2022).In addition, during AD development, microglia acquire an AD signature; for example, many AD risk genes are highly enriched in microglia, some of which are exclusively expressed in microglia.These include apolipoprotein E (APOE),triggering receptor expressed on myeloid cells 2 (TREM2), ATP binding cassette subfamily A member 7 (ABCA7), complement C3b/C4b receptor 1 (CR1), Spi-1 proto-oncogene (SPI1), and MS4A-related family (MS4As)(Bertram et al., 2008; Efthymiou and Goate, 2017), highlighting the important role of microglia in sporadic AD.
In addition to these important genetic risk factors, environmental risk also plays a role that should not be underestimated.Up to one-third of patients with AD may be affected by modifiable risk factors (Livingston et al., 2017).Environmental factors may affect microglia, and microglia may in turn affect an individual’s susceptibility to AD, as microglial dysfunction is a major risk factor for AD (Bisht et al., 2018; Katsumoto et al., 2018; Phan and Malkani,2019).Stress during both perinatal and adult development can lead to microglial function changes, which in turn affect cognitive function (Tay et al.,2017).Specifically, particulate matter (PM) with diameter < 2.5 µm (PM2.5) can reach the CNS through various routes and there activate microglia to cause inflammation and nerve damage, leading to AD (Shi et al., 2020).Dietary factors can modulate gut microbiota composition, and the gut microbiota may be associated with AD pathogenesis by altering microglial function (Bairamian et al., 2022).Circadian rhythm disturbance and other poor lifestyle factors cause microglia neurodegeneration (Phan and Malkani, 2019).While neither genetic risk factors nor environmental exposures affect AD alone, together they may interact to promote AD progression; however, those interactions are poorly understood (Dunn et al., 2019).Almost all environmental exposures described herein may interact with the APOE genotype, and thus affect cognitive function (Rajan et al., 2021).Herein, we discuss the impacts of environmental risk factors (e.g., air pollution) and modern lifestyles (e.g., chronic stress, poor eating habits and circadian rhythm disturbance, inadequate physical exercise) on AD pathogenesis and progression, by contributing to pathogenic microglial phenotypes and modulating their functions.These cumulative findings support the idea that microglial dysfunction is a central mechanism of AD development and pathology progression.The positive roles of healthy environment and lifestyle factors are discussed in relation to reducing individual susceptibility to AD, and their modulation of microglial function is considered as a potential future therapeutic modality.
Retrieval Strategy
An online PubMed search was performed, retrieving articles published through August 31, 2023.Combined MeSH terms were used to maximize search specificity and sensitivity: “Alzheimer’s disease”; “microglia”; “environmental pollution”; “chronic stress”; “gut microbiota”; “PM2.5”; “heavy metals”; “sleep”;“smoking”; “physical exercise,” and “coffee.” The results were further screened by title and abstract, and only studies exploring the interplay between microglia and environmental AD risk factors were included, to facilitate the goal of reviewing the effects of environmental risk factors on microglia characteristics in AD.No language or study type restrictions were applied.
Environmental Exposure Contributions to Alzheimer’s Disease Risk
Air pollution
Air pollution, a complex mixture of substances, is a global environmental problem that kills 7 million people annually worldwide (Colao et al., 2016).Air pollution contains PM, ozone, nitrogen dioxide, and other substances.Among these, the most harmful to our health is ambient fine particulate matter PM2.5(Choi et al., 2018).There is a strong link between PM2.5and cognitive dysfunction, including AD (Thiankhaw et al., 2022).Studies have also verified how PM2.5enters the CNS, including via both the blood-brain barrier (BBB)and olfactory neurons (Ajmani et al., 2016).The BBB is a highly selective semipermeable membrane that restricts the flow of many substances into the brain and plays an important role in maintaining CNS homeostasis (Alahmari,2021).In animal models, PM-mediated tight junction protein expression leads to reduced BBB integrity and increased permeability (Oppenheim et al., 2013).In addition, the olfactory nerve is considered a critical pathway by which PM2.5affects the CNS (Li et al., 2022).Rodent models have confirmed that PM can access the brain via the nose (Elder et al., 2006).Considerable PM parts can be deposited in the nasopharyngeal region, to be translocated further into multiple brain regions, circumventing the tight BBB (Oberdorster et al., 2004).By entering the CNS through these two main pathways, PM2.5exposure increases AD risk.However, the exact mechanism and causal relations between PM2.5and AD remain unclear, requiring further research.
PM2.5 exposure
Investigators increasingly show that long-term PM2.5exposure higher than that specified by United States Environmental Protection Agency (US EPA)standards may be related to cognitive decline and accelerated brain ageing(Kang et al., 2021; Lee et al., 2021; Patten et al., 2021).Observational studies in children (Calderon-Garciduenas et al., 2015), older adults (Ailshire and Crimmins, 2014), and dogs (Calderon-Garciduenas et al., 2008b) living in areas with higher PM2.5concentrations have indicated that this prolonged exposure can induce neuroinflammation, increase AD risk, and accelerate AD pathogenesis.Some of the AD-linked molecular and cellular alterations caused by PM2.5include mitochondrial dysfunction, oxidative stress, microglial activation, neuroinflammation, synaptic dysfunction, and neurovascular dysfunction (Gonzalez-Maciel et al., 2017; Wang et al., 2020a).PM2.5can also alter major AD markers, including elevated levels of Aβ accumulation, tau hyperphosphorylation, and misfolded α-synuclein (Cheng et al., 2016; Costa et al., 2017).
Current clinical studies have shown a clear correlation between environmental air pollution and AD pathology, based on three methods: biomarkers,neuroimaging, and epigenetics.The first line of evidence has shown that in the cerebrospinal fluid of children and young people living long-term in areas where annual average PM2.5concentrations exceed the US EPA standard level long-term, Aβ and brain-derived neurotrophic factor (BDNF) are reduced, and tau protein, inflammation, and cytokine marker concentrations are increased(Calderon-Garciduenas et al., 2018).The second line of evidence indicates that patients with mild dementia or MCI who live in areas with higher PM2.5concentrations have a higher likelihood of amyloid PET scan positivity(Iaccarino et al., 2021).The third line of evidence indicates close relations between low deoxyribonucleic acid (DNA) methylation and AD in both epidemiological and experimental studies (Bakulski et al., 2012; Chouliaras et al., 2013; Wei et al., 2020).Researchers have speculated that PM2.5can reduce genome-wide DNA methylation or global hypomethylation, including of BACE1, APOE, and APP (Shou et al., 2019).In summary, effects of PM2.5on AD can be detected by cerebrospinal fluid biomarkers, DNA methylation levels, and amyloid deposition on PET scans.
PM2.5-promoted microglial activation
Since microglia-mediated neuroinflammation is a detrimental AD event,researchers have assessed whether and how PM2.5affects microglia function.Previous research used diesel exhaust particles (DEPs) phagocytosed by microglia to produce superoxide, showing that selective dopaminergic neurotoxicity occurred only in the presence of microglia, indicating that microglia-derived reactive oxygen species (ROS) are crucial for DEPinduced dopaminergic neurotoxicity (Block et al., 2004).This finding supported the idea that DEPs are culpable in microglial activation (Block et al.,2004).Moreover, prolonged exposure to high levels of air pollution leads to increased expressions of CD14 (Calderon-Garciduenas et al., 2008a) and the microglial marker Iba1 (Bai et al., 2019), indicating that microglia may be the main targets of air pollution.
Anin vitroexperiment showed that DEP exposure affected microglia in various ways, including cytotoxicity, oxidative stress, lipid peroxidation,neuroinflammation, activation, autophagy, and apoptosis (Bai et al., 2019).PM2.5significantly increased the level of light-chain 3b (LC3b) in microglia,resulting in a DAM microglia phenotype representing enhanced phagocytosis(Kang et al., 2021).A recent study suggested that PM2.5decreases microglial viability and promotes microglial activation.Specifically, PM2.5increased the introduction of proinflammatory molecular markers (i.e., tumor necrosis factor alpha [TNF-α], interleukin-6 [IL-6], interleukin 1 beta [IL-1β], inducible nitric oxide synthase [iNOS], and COX-2) while inhibiting the development of anti-inflammatory molecular markers (IL-10 and Arg-1) (Kim et al., 2020).These data further indicate that PM2.5mediates AD pathogenesis through microglia-produced neuroinflammation and oxidative stress.PM2.5is now widely studied as a cause of neuroinflammation.Exposure to PM2.5decreases the activity of several antioxidant enzymes that scavenge cellular free radicals and protect cells, including superoxide dismutase, malondialdehyde, heme oxygenase-1 (HO-1), and GSH-Px, while increasing brain ROS (Herr et al., 2021;Zhu et al., 2023).One mechanistic study showed that PM2.5can downregulate miR-574-5p, which targets BACE1 through nuclear factor kappa B (NF-κB) p65,thereby causing neuroinflammation, disrupting synaptic functional integrity,and accelerating cognitive dysfunction (Ku et al., 2017).Another mechanistic study showed that PM2.5exposure activates the microglial HMGB1-NLRP3-P2X7R signaling pathway and reduces hippocampal neuron viability.That group also found that HMGB1-NLRP3 pathway downregulation inhibited microglial activation, decreased inflammatory factor production, and restored hippocampal neurons functioning (Deng et al., 2022).
Thought various opinions exist regarding how PM2.5accelerates AD via microglia, and no consensus has formed.PM2.5may thus directly or indirectly activate microglia through three ways.First, by directly activating microglia.Second, by activating proinflammatory signals in peripheral tissues or organs,including the liver (Jeong et al., 2019), lung (Jia et al., 2021) and cardiovascular system (Wang et al., 2015), eliciting a circulating cytokine response (Ruckerl et al., 2007) which activates microglia and causes neuroinflammation (Block and Calderon-Garciduenas, 2009).And third, by directly damaging neurons to activate microglia (Liu et al., 2021).However, given the complexity of PM2.5exposure, it is unclear whether it activates microglia through one of these modalities or in combination among them in AD pathological conditions.The evidence for the influences of PM exposure on microglia are summarized in
Table 1.
To date, animal model experiments, epidemiological studies, and clinical studies have confirmed a causal link between ambient air pollution and AD pathology.The underlying mechanism for this may be that PM2.5activates microglia via one or more of the three pathways described above, thereby inducing neuroinflammation and oxidative stress and leading to AD biomarker changes (Figure 1).In addition to microglia, PM2.5can also act directly on other neural cells.We previously reported that PM2.5can cause direct neuronal damage under very short-term exposure, whereas microglia did not play a major role (Liang et al., 2023).However, changes in microglia in AD under long-term exposure to PM2.5require further exploration.

Figure 1 |Possible mechanisms by which microglia detect PM2.5 and some ADrelated neuropathologic consequences of this exposure.

Table 1 |Summary of known instances of PM exposure that influence microglia
Heavy metals: powerful AD risk factors
Among environmental factors, the contributions of heavy metals such as lead(Pb), cadmium, and manganese to AD is of interest, given the wide range of their population-level exposures.The effects of various common heavy metals on AD have been well studied (Plascencia-Villa and Perry, 2021).For example, childhood Pb exposure has been shown to lead to memory loss and cognitive decline in older age (Reuben et al., 2017).In vitrostudies have shown that Pb exposure increases the water content of tau in SH-SY5Y cells(Bihaqi et al., 2017).Different metals, such as Pb, arsenic, and cadmium,may induce Aβ production to the greatest extent through synergistic effects(Ashok et al., 2015).However, whether the respective mechanisms are the same is still poorly understood (Islam et al., 2022).Some metal chelators,such as hydroxypyridones, can interfere with protein folding and prevent its undergoing oxidative processes (Singh et al., 2019).Therefore, metal chelator studies may facilitate development of novel anti-AD agents as potential therapies (Fasae et al., 2021).In summary, adverse environmental factors lead to the phenotype of pathogenic microglia and regulate their function, thereby increasing individuals’ susceptibility to AD.
Stress
Though stress is an inevitable part of life, it can also endanger human health under certain conditions.Given constantly changing, fast-paced modern lifestyles, stress is a frequent conversation topic (McEwen, 2005).Stress is divided into acute and chronic.The effects of acute stress can be quickly alleviated, after which the condition normalizes.However, chronic stress,such as long-term emotional stress, results in homeostatic dysregulation that can lead to various diseases, especially in susceptible individuals (Saeedi and Rashidy-Pour, 2021).These include, but are not limited to, mental illness (Menard et al., 2017), cardiovascular diseases (Franklin et al., 2021),gastrointestinal disorders (Alonso et al., 2008), and neurodegenerative diseases (Sazonova et al., 2021).Though stress can increase the occurrence,and worsen the symptoms, of AD, the most common neurodegenerative disease, the mechanism for this relation is as yet unclear.
Stress and cognitive decline: epidemiology and pathology evidence
Growing evidence recognizes chronic psychosocial stress as a risk factor for late-onset AD, and a strong contributor to promoting AD brain pathology(Gracia-Garcia et al., 2015; Piirainen et al., 2017).Epidemiological, clinical,and animal studies suggest that chronic uncontrollable and unpredictable stressors are associated with mental illnesses, including depression and neurodegenerative disorders like AD (Liu et al., 2017).Epidemiological studies have found that depression is an AD risk factor, and that people who are prone to psychological stress are at higher risk for AD (Wilson et al., 2003; Alkadhi,2012).Chronic psychosocial stress is increasingly considered a risk factor for late-onset AD and its related cognitive impairment (Saeedi and Rashidy-Pour, 2021).Compared with healthy age-matched controls, patients with AD present increased peripheral and CNS cortisol levels, reflecting a relation between stress and AD (Popp et al., 2015).In animal experiments, acute stress (i.e., acute cold water stress) (Feng et al., 2005), acute unpredictable stress (Filipcik et al., 2012) and chronic stress (i.e., chronic restraint stress)(Yan et al., 2010), and chronic unpredictable mild stress (Briones et al.,2012) all increase tau phosphorylation.Similar to the consequences of tau phosphorylation, female 5×FAD mice exposed to chronic restraint stress in the prepathological stage exhibited elevated BACE1 and APP levels, increased neurotoxic Aβ42levels, and hippocampal plaque deposition (Devi et al., 2010;Ray et al., 2011).It has been demonstrated that chronic stress can promote the emergence of AD and accelerate its progression through synaptic loss and impaired neurogenesis (Xie et al., 2021).For example, social isolation accelerated AD onset and spatial working memory impairment in adult APP/PS1 transgenic mice (Huang et al., 2011).Overall, these studies suggest that stress can exacerbate and accelerate AD progression.
Effects of stress on microglia: a double-edged sword?
Although the mechanism is not yet clear, microglia are suspected to play a significant role in promoting AD development and progression under chronic stress.Chronic stress may activate microglia, induce inflammation, and worsen cognitive function in the adult brain (Piirainen et al., 2017).In the hippocampus of stressed rats, Iba1 and CD11b protein levels were significantly increased compared with those of non-stressed control rats; thus, chronic mild stress induced microglial proliferation in the hippocampus (Du Preez et al., 2021).Chronic stress also selectively increases the density of microglia in certain stress-sensitive brain regions, such as the prelimbic cortex (Han et al.,2020), medial prefrontal cortex (Bollinger et al., 2016), and hippocampal CA3 region (Bian et al., 2012).Quantification of microglia from mice subjected to restraint stress indicate that augmented density is caused by proliferation(Nair and Bonneau, 2006).Although these studies imply that chronic stress can stimulate microglial proliferation, the mechanism of action is poorly understood.In a Chinese herbal medicine study, chronic stress modulated microglia toll-like receptor 4 (TLR4)-I-kappa B kinase-NF-κB signaling pathway, promoting the release of inflammatory factors from hippocampal microglia and leading to neuroinflammation (Qu et al., 2021).Nrf2-HO-1-NLRP3 signaling was inhibited in microglia exposed to stress using Nrf2 siRNA transfection, thereby promoting microglial polarization to M1 and inhibiting microglial polarization to M2 (Tao et al., 2021).More mechanisms are under investigation, the discovery of which will facilitate new AD therapeutics.
One urgent question is whether chronic stress alters microglial morphology.As it is tightly coupled to its function, using different stress models had led to varying results.Chronic stress causes a marked transition of microglia from a ramified-resting state to a non-resting state.A primary microglial morphological change observed in the chronic stress paradigm is a hyperramification state, with longer and more branched processes (Hinwood et al., 2013; Hellwig et al., 2016).This suggests that chronic stress increases microglial structural complexity but does not make them larger.Another main microglial change is cellular deramification, with increased soma size and shortened processes (Kreisel et al., 2014; Wohleb et al., 2014).However,Lehmann and colleagues found that neither acute nor chronic social defeat stress changed microglial morphology, including cell perimeter length, cell spread, eccentricity, roundness, or soma size (Lehmann et al., 2016).These cumulative results indicate that there is currently no consensus regarding the effects of stress on microglial morphology.
In the mature healthy brain, microglia remove cellular debris and necrotic or apoptotic cells through phagocytosis.Several investigators have investigated whether exposure to stress modulates the phagocytic function of microglia.Milior et al.(2016) exposed mice to a chronic stressor and examined hippocampal CA1 radiatum with electron microscopy, to assess the number of phagocytic inclusions per Iba1-positive microglial process.They found that exposure to stressful (versuscontrol) environments increased the microglial inclusion index.Similarly, enhanced microglial phagocytic capacity was found in Thy-1-GFP transgenic mice subjected to a 14-day chronic unpredictable stress (CUS) model (Wohleb et al., 2018).These results indicate that CUS increases the microglial phagocytosis of neuronal components.Lehmann et al.(2016) also replicated this phenomenon with chronic, but not acute social defeat.Thus, stress may induce neuronal remodeling by enhancing microglial phagocytic function.
In recent decades, efforts have been made to determine how stresses alter microglia activity of (Frank et al., 2019; Woodburn et al., 2021).Although the specific signaling pathways by which stress activates microglia are unclear (Wohleb et al., 2016), a growing body of evidence implicates several factors, such as cytokines, pattern recognition receptor agonists, and stress hormones.First, exposure to stressors releases pathogen-associated and danger-associated molecular patterns into the bloodstream, which can trigger microglia and amplify neuroinflammatory responses (Wenzel et al., 2020).Second, the hypothalamic-pituitary-adrenal (HPA) axis plays an important role in stress-induced microglial activation (Frank et al., 2012).Stress can initiate proinflammatory responses in microglia by activating the HPA axis, releasing more corticotropin-releasing hormone and glucocorticoids (GCs) (Van den Bergh et al., 2020).GCs are a suspected link between chronic stress and altered microglial structure and function.Moreover, both pharmacological(glucocorticoid receptor [GR] antagonist RU486) and surgical (adrenalectomy)treatments block stress-induced microglial priming (Frank et al., 2012)and reduce markers of phagocytosis on petrified microglia following CUS(Pedrazzoli et al., 2019).These cumulative studies demonstrate that GCs may play a pivotal role in stress-induced priming of microglial activation.Although the mechanism of cognitive impairment caused by chronic stress is unclear,several GR-regulated genes are closely related to AD.First, expression of GRregulated gene Dusp1, which phosphorylates tau kinase, is decreased during chronic stress, leading to tau hyperphosphorylation.DUSP1 expression is also decreased in the brains of AD mice compared with age-matched controls(Arango-Lievano et al., 2016).Another GR-regulated gene, Sgk1, a key modifier of tau pathology in AD, is upregulated in the brains of participants with AD.Sgk1 inhibition reduces tau neuropathology and improves cognition in preclinical AD mouse models (Elahi et al., 2021).
Chronic stress is a modifiable AD risk factor.This relation may be caused by HPA axis dysregulation and other molecular mechanisms, which can lead to a series of microglial changes.Under chronic stress, elevated GC levels prompt microglial proliferation and enhance their phagocytic ability, though there is no uniform conclusion regarding the morphological effects.These changes allow microglia to undergo a notably increased proinflammatory response, leading to increased neurotoxic cytokines, accumulating Aβ, and tau.These combined factors may accelerate AD onset (Figure 2).Determining the underlying mechanism of the association between chronic stress and AD may help develop new therapeutic targets and chronic stress management options.
Gut Microbiota
There is a close homology among animals, including humans, in their microbial communities, which are comprised of fungi, archaea, bacteria,and viruses.Gut microbiota, which make up the gastrointestinal community,represents the greatest density and absolute abundance of microorganisms in the human body.Throughout the human life span, the gut microbiota is a dynamic, diverse community that may change in response to both extrinsic and host factors, such as drugs (Vich Vila et al., 2020), diet (David et al., 2014),age, and sex (de la Cuesta-Zuluaga et al., 2019).The microbiota-gut-brain axis,a complex bidirectional communication system between the brain and gut microbiota, is mediated by direct and indirect signaling, including nervous,endocrine, and immune mechanisms (Martin et al., 2018).The gut microbiota is a critical regulator within the microbiota-gut-brain axis, sensing, modifying,and tuning chemical signals from the brain.It may also affect AD pathogenesis and cognitive decline.
Gut microbiota in AD: positive progress and unclear mechanism
Researchers recently identified a strong correlation between altered gut microbiota and cognitive performance (Hu et al., 2016).It has also been found that microbiota composition and diversity are perturbed in AD mice(Fox et al., 2019), implying that the microbiota has a nonnegligible role in AD progression.Compared with control participants, gut microbiome microbial richness and diversity were both decreased in participants with AD (Jung et al., 2022).The abundance of phylum-level Firmicutes and Actinobacteria is lower, and the abundance of Bacteroidetes higher, in those with AD (Vogt et al., 2017).At the family level, Helicobacteraceae, Coriobacteriaceae,and Desulfovibrionaceae are significantly more abundant in APP/PS1 mice compared with wild-type (WT) mice.At the genus level, the mean abundances of Odouribacter and Helicobacter in APP/PS1 mice are obviously higher compared with WT mice, and Prevotella is significantly higher in WT compared with APP/PS1 mice (Shen et al., 2017).However, 16S rDNA sequencing showed gut microbiota diversity increases in AD Drosophila.At the family level, the proportions of Acetobacteraceae and Lactobacillaceae in AD Drosophila decreased dramatically, although they were largely enriched in the Drosophila microbiota.At the genus level, the mean control group abundances of Acetobacter and Lactobacillus were significantly higher compared with AD flies (Kong et al., 2018).Furthermore, metabolites derived from the gut microbiota, such as trimethylamine N-oxide, are elevated in the cerebrospinal fluid of individuals with AD, and elevated trimethylamine N-oxide is associated with AD pathology (Vogt et al., 2018).Despite significant differences and variation in gut microbiota among AD model species, these studies suggest a close relation between disturbances in gut microbiota and AD (Table 2).
Despite the complex and numerous gut microbiota, several mechanisms for its involvement in, and impact on, AD have been explored.Asparagine endopeptidase (AEP), also known as delta-secretase, can simultaneously cleave increased APP and tau in the brain to form amyloid plaques and neurofibrillary tangles (Chen et al., 2021).In addition, enhancer-binding protein beta (C/EBPβ) is an Aβ and proinflammatory cytokine-activated transcription factor that regulates AEP transcription and protein levels in an age-dependent manner (Wang et al., 2018b).In the brains of mice with fecal transplant from patients with AD, investigators have found that C-EBPβ-AEP signaling is significantly activated and that mRNA transcription of major enzymes involved in arachidonic acid (AA) metabolism is increased.Elevated enzymes lead to upregulation of various AA metabolites, including prostaglandin E2 receptor EP3 subtype-like (PGE2), thromboxane B2, LKB4,and 12-HHT, among others, which stimulate microglial activation to aggravate neuroinflammation (Chen et al., 2022).EBPβ regulates proinflammatory genes in glial cells, such as nitric oxide synthase 2, IL-1β, IL-6, and TNF-α(Straccia et al., 2011).The most direct route may be that gut microbiota in the feces of patients with AD directly upregulates C-EBPβ-AEP signaling, and APP and tau are cleaved by activated AEPs, directly triggering AD pathogenesis (Xu et al., 2023).Hypothesized relations between intestinal microbiota imbalance and AD pathology, as well as the repair of the microbiota-gut-brain axis by antibiotic therapy and probiotic supplements, are shown in Figure 3.
Several classic interventions have been used to assess the relations between the gut microbiota and AD, including probiotic or antibiotic treatments,establishment of sterile animals, and fecal microbiota transplantation.These have demonstrated cognitive function improvements among patients with AD after 12-week probiotic consumption (Akbari et al., 2016).In 3×Tg-AD mice,early-stage AD progression was modulated by SLAB51 probiotic formulation(Bonfili et al., 2017).In a rat AD model, Morris water maze indicated beneficial effects of probiotics on cognitive function (Bonfili et al., 2017).Continuous ingestion of probiotics and prebiotics have been showed to delay neurocognitive decline and reduce AD risk (Tillisch et al., 2013; Akbari et al.,2016; Abraham et al., 2019; Kobayashi et al., 2019).Furthermore, a long-term broad-spectrum combined antibiotic regimen decreased Aβ plaque deposition and improved cognitive functions in a murine AD model (Minter et al., 2016).In rodent AD models, antibiotics had similar effects on AD pathogenesis, and reduced microglial activation, inflammatory cytokines, and brain Aβ (Yulug et al., 2018; Zhao et al., 2022).Microbiota transplantation from aged APP/PS1 mice significantly increased Aβ in germ-free (GF) APP/PS1 mice (Harach et al., 2017).Additionally, a phase 3 clinical trial in China found that the sodium oligomannate GV-971 inhibits intestinal microecological disorders and related phenylalanine/isoleucine accumulation, controls neuroinflammation, and reverses cognitive impairment (Wang et al., 2019; Xiao et al., 2021).These cumulative studies support a close relation between gut microbiota and AD;while the specific mechanism remains unclear, neuroinflammation may be an important link.

Table 2 |Variation in gut microbiota in Alzheimer’s disease (AD) patients and AD models
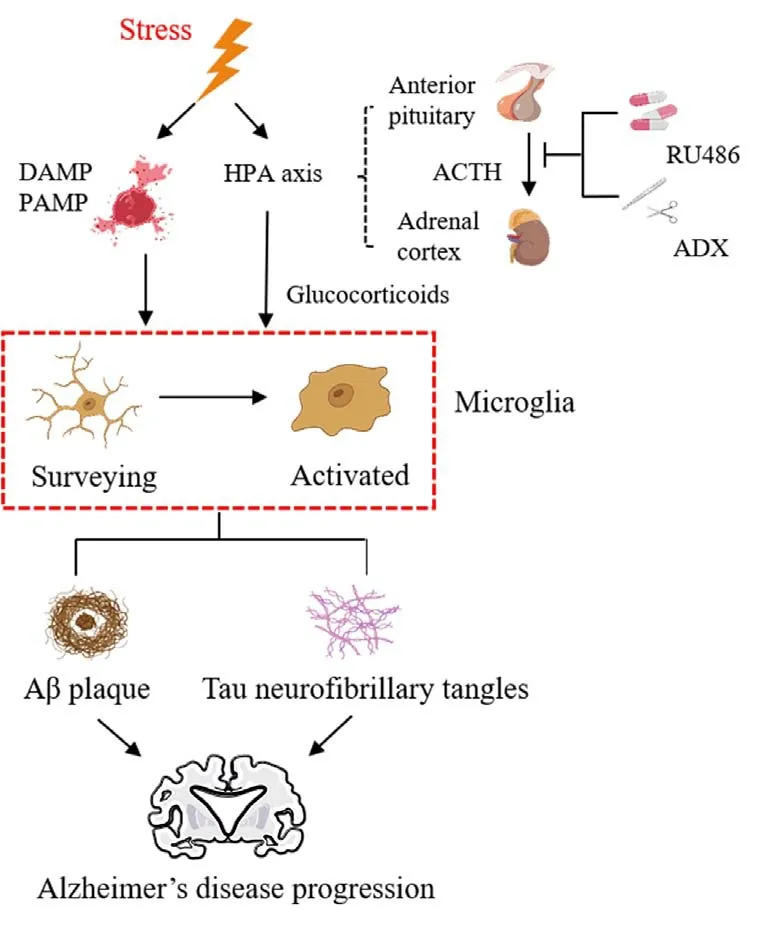
Figure 2 |Microglia as stress response sensors.

Figure 3 |Postulated links between gut microbiota dysregulation and AD pathology,and microbiota-gut-brain axis reconditioning with antibiotic treatment and probiotic supplementation.
Microbiota and microglia homeostasis
Given that the gut microbiota is an essential regulator of microglial function,microbiota-microglia interactions may be a critical link to CNS disorders.Observations from GF animals (which are hand-raised in an aseptic isolator without microorganism exposure) indicate important contributions of the gut microbiota to microglial homeostasis, including global defects from an immature phenotype, altered steady-state conditions, and diminished immune responses.When Erny et al.(2015) compared microglial morphology characteristics between GF and specific pathogen-free mice,microglia of the former had more complex morphologies, including longer processes, more segmented branches and terminal points, and greater density.Depletion of intestinal bacteria by antibiotic treatment induced a microglia phenotype comparable to that of GF mice.Microglial immaturity and malformation were restored by reintroducing complex, live microbiota or microbial metabolites such as short-chain fatty acids (Erny et al., 2015).Notably, the double transgenic APP/PS1 AD mouse model housed under GF conditions showed lower brain Aβ levels and deposition compared with APP/PS1 mice (Harach et al., 2017).
These studies emphasize the indispensable role of gut microbiota in microglia maturation and function, raising the possibility that microglia may require continuous input from the host microbiota to maintain CNS homeostasis.The mechanism by which unhealthy gut microbiota activates microglia remains to be determined.Close relations between potassium channels KCa3.1 and Kv1.3 and microglial activation have been shown repeatedly (Cocozza et al., 2021).Levels of KCa3.1 and Kv1.3 and mRNA levels of related inflammatory factors were increased in microglia isolated from brains of Western diet-fed mice(Jena et al., 2018).Additionally, Western diet-fed-derived saturated fatty acid can activate the microglial CD14-TLR4-MD2 complex, ultimately damaging microglia by inducing NF-κB signaling, which triggers neuroinflammation and proinflammatory cytokine secretions (Wieckowska-Gacek et al., 2021).
In summary, clinical and animal studies support relations between the gut microbiota and AD.While the specific underlying mechanism remains unknown, neuroinflammation caused by microglia may be an important contributor.According to current research, the classic interventions described above impact intestinal flora composition, and thus affect host cognitive behavior.Therefore, an in-depth understanding of the relations between intestinal flora and AD may open new methods for early AD treatment.
Other lifestyles factors linking microglia and AD
In addition to the aforementioned risk factors, AD is driven by numerous others, emphasizing the disease’s complex, multifactorial nature.The evidence supporting several of these factors is described here.
Sleep and circadian rhythmicity
Sleep plays an important role in cognition and memory.Sleep disturbance(SD) increases Aβ burden, potentially triggering cognitive decline and increasing AD risk (Irwin and Vitiello, 2019).Microglial activation is a key mediator of SD-induced imbalance in inflammatory cytokines and cognitive impairment (Wadhwa et al., 2017a), and inhibiting microglial activation with minocycline can intervene to improve cognitive performance during SD(Wadhwa et al., 2017b).Furthermore, microglia possess a circadian clock that influences inflammatory responses via robust rhythms of TNF-α, IL-1β, and IL-6 mRNA (Fonken et al., 2015).In addition to inflammatory factor releases,other microglia functions are similarly influenced by circadian rhythms.One circadian clock component, Rev-erbα, plays a key role in microglial activation and neuroinflammation (Griffin et al., 2019).The Rev-erbα agonist SR9011 switches microglia to a neuroprotective phenotype (Griffin et al.,2020), decreasing phagocytic microglial capacity (Wang et al., 2020c), and inhibiting cellular metabolism (Wolffet al., 2020).Therefore, circadian rhythm disturbances may also affect microglial function by modulating the basic helixloop-helix ARNT like 1 (BMAL1)-REV-ERBα axis, which can lead to cognitive dysfunction.
Physical exercise
The neuroprotective roles of physical exercise have been confirmed in several studies (Chen et al., 2016).It is well known that regular physical exercise is a modifiable lifestyle factor that can reduce AD risk and slow its progression.The functions of regular physical activity include cognitive improvement and amelioration of Aβ deposition and tau phosphorylation (Kelly, 2018).Compared with aged controls without neurological disease, postmortem brain samples from patients with AD show higher levels of proinflammatory microglia and lower levels of anti-inflammatory microglia (Kohman et al.,2013; He et al., 2017; Jiang et al., 2017).Physical exercise also increases the number of anti-inflammatory phenotype microglia and suppresses activation of proinflammatory phenotype microglia in the hippocampus (Zhang et al., 2019), which are regulated by increasing anti-inflammatory factors and suppressing the production of proinflammatory cytokines and chemokines (Lu et al., 2017; Mee-Inta et al., 2019).Therefore, exercise may regulate microglia function and induce anti-inflammatory effects.
Smoking
Cigarette smoking is the most preventable cause of death and various diseases, including cardiovascular disease, lung disease, and various cancers(Billatos et al., 2021; Fagerberg and Barregard, 2021; Virani et al., 2021; Hecht and Hatsukami, 2022).Smoking not only increases the incidence of these diseases, it leads to neurocognitive abnormalities, significantly increasing symptom progression over time (Durazzo et al., 2010).Epidemiology studies have explored several aspects of the association between smoking and AD.First, the likelihood of AD in smokers is greatly increased; second, smokers have a lower age of AD onset compared with nonsmokers; and third,decreasing smoking prevalence may reduce the future AD incidence (Barnes and Yaffe, 2011; Henderson, 2014).To some extent, smoking harms the brain in ways similar to that of PM2.5 or other air pollution.Cigarette smoking causes brain alterations including increased oxidative stress (Durazzo et al.,2016b), decreased hippocampal and lefthippocampal volumes (Durazzo et al., 2013), and inhibited dentate gyrus neurogenesis (Durazzo et al., 2016b).In terms of pathological changes, cigarette smoking tends to be associated with more amyloid deposition and tau phosphorylation (Durazzo et al.,2016b), and lower rates of cortical glucose metabolism have been shown in autopsy and clinical studies (Durazzo et al., 2016a).Thus, while smoking is a major modifiable risk factor for AD, the underlying mechanisms are still being investigated.Because one possible mechanism is neuroinflammation resulting from microglial activation, our group explored microglia affected by chronic cigarette smoke (CS).Quantitatively, animal studies have shown that long-term chronic CS exposure can induce neuroinflammation by increasing microglial activation, which can be reduced following acute nicotine withdrawal (Adeluyi et al., 2019; Prasedya et al., 2020; Sivandzade et al.,2020).Morphologically, CS transforms hippocampal microglia into amoeboid shapes, mainly characterized by a decrease in cell processes total length and mean branch numbers in the CA3, but not the CA1 or dentate gyrus regions(Dobric et al., 2022).Thus, chronic CS exposure causes active morphologic changes, and specifically reducing microglial numbers.In conclusion,understanding how smoking impacts cognition by altering microglia may aid developing microglial modulators as a smoking-induced AD treatment.This remains an area in which significant research is needed.
Caffeine
Coffee, among the most widely-consumed beverages worldwide, is correlated with reduced AD risk (Londzin et al., 2021).The mechanisms of caffeine in AD prevention and regulation include: (1) Reduced Aβ plaque production via decreased expressions of presenilin 1 (PS1) and β-secretase (BACE), a possible mechanism by which caffeine protects cognition (Arendash et al.,2006); (2) Reduced phosphorylation levels by reducing glycogen synthase kinase 3 alpha dysregulation and glycogen synthase kinase 3 beta expression,both of which result from attenuating TNF-α-c-Jun and Akt-mTOR signaling(Arendash et al., 2009; Zhou and Zhang, 2021); and (3) Attenuation of AD-associated neuroinflammation by inhibiting the excessive microglia activation and reducing BBB disruption (Madeira et al., 2017).In onein vivostudy, daily injection of caffeine for 4 weeks significantly decreased Iba-1 expression and microglial number in the LPS-treated mouse brain, though morphological changes were not significant (Badshah et al., 2019; Yang et al.,2022).It has also been demonstrated that caffeine decreases the microglial proinflammatory phenotype by downregulating the levels of CD86 and iNOS and inhibiting TNF-α and IL-1β secretions.In contrast, caffeine upregulates the anti-inflammatory phenotype of microglia, elevates expressions of CD206 and Arg1, and promotes secretion of anti-inflammatory factors (Yang et al., 2022).Caffeine inhibits microglial hyperactivation, reduces microglial polarization to the proinflammatory phenotype, and promotes microglial polarization to the anti-inflammatory type (Yang et al., 2022).Furthermore, pKr-2 induces hippocampal neurodegeneration by stimulating TLR4 with PU.1 and c-Jun,serving as a proinflammatory stimulus for microglial response.Caffeine also inhibits hippocampal pKr-2 expression and reduces TLR4 upregulation in 5xFAD mouse microglia, alleviating hippocampal neurodegeneration (Kim et al., 2023).While it is therefore reasonable to speculate that the positive effects of caffeine on AD are achieved via shifts in microglial phenotype, the specific mechanism is currently unclear.
Limitations
Some limitations to this review should be acknowledged.Due to the unique nature of brain diseases, the research findings summarized herein regarding how modern lifestyle factors such as air quality, stress, gut microbiota, sleep,physical exercise, smoking, and caffeine regulate microglia and AD are largely based on cellular or rodent model studies.However, these findings remain to be confirmed by epidemiological and clinical data.Several methods found to slow AD progression by microglia-based intervention (e.g., microglia ablation,microglia inhibitors, gene knockout, stem cell transplantation) also require further evaluation to facilitate clinical research.
Conclusions and Perspectives
Age is an independent risk factor for AD, a multifactorial disease affected by both modifiable environmental risk factors and genetics.Herein, we reviewed how modern lifestyle factors, including air quality (i.e., PM2.5), stress, the gut microbiota, sleep and circadian rhythms, and physical exercise, affect animal models and human microglia regulation in the AD context.The exact role and importance of microglia in the onset and development of AD is a current research hotspot, as is how different lifestyle factors regulate microglia and affect AD.Notably, these lifestyle factors do not affect AD via microglia independently.For example, chronic sleep disruption affects the gut microbiota by altering taxonomic profiles of fecal microbiota.And exercise is also involved in the regulation of circadian melatonin rhythm and the sleep-wake cycle (Tahara and Shibata, 2018).Various environmental and lifestyle factors may thus synergistically promote AD development.Increased AD susceptibility can be driven by modulation of microglia through different lifestyles above, thus determining the true role and importance of microglia in AD remains an area worthy of further investigation at present.
Clinical pharmacology now used in AD treatment generally only addresses symptoms, rather than preventing the disease (Cai et al., 2022).The hypothesis that microglial activation may adversely affect disease progression has prompted the search for ways to deplete microglia via compounds and genetic models.Although various methods are available for microglial ablation, the use of most depletion methods, including the CX3CR1creERxDTRffmouse model (Bruttger et al., 2015; Rice et al.,2017), the CD11b-HSVTK model (Grathwohl et al., 2009), and clodronate liposomes (Faustino et al., 2011) is prohibited for various reasons.In contrast to microglial depletion paradigms, CSF1R inhibitors have unique advantages due to their highly selective effects, noninvasive administration, and rapid/sustained microglia elimination.CSF1R inhibitors PLX5622 and PLX3397 eliminate almost all microglia from the 5xFAD mouse brain and prevent plaque formation (Spangenberg et al., 2019), concomitant with dopaminergic signaling rescue (Son et al., 2020).
Blockage of microglial activation is neuroprotective in AD animal models(Akiyama et al., 2000).The beneficial effects of nonsteroidal anti-inflammatory drug treatment have been examined in multiple epidemiological and animal model studies (McGeer and McGeer, 2013).Numerous epidemiological studies have indicated that nonsteroidal anti-inflammatory drug use may protect against AD development, though this result is not clinically significant and its mechanism of action is unclear (Etminan et al., 2003; Rosenberg,2005; Ozben and Ozben, 2019).Minocycline, a commonly used tetracycline antibiotic, is a strong inhibitor of the microglial shift to a proinflammatory phenotype (Kobayashi et al., 2013).As a modulator of microglial inflammatory responses, minocycline has also shown neuroprotective effects in an AD animal model (McLarnon, 2019).
As described above, genome-wide association studies have identified many risk factors expressed by microglia in AD.Ablation of the NLRP3 gene is protective in AD models.Using an AD mouse model of APP/PS1, Aβ pathology and Aβ-induced synaptic damage were markedly reduced in Nlrp3 knockout mice (APP/PS1/Nlrp3-/-) (Scheiblich et al., 2020).In addition to knockouts,several NLRP3 inhibitors have been described, including the sulfonylureacontaining compound MCC950.In vivo, this compound can inhibit ATP hydrolysis and IL-1β release, reduce amyloid plaque burden, and improve cognitive behavior in AD model mice (Thawkar and Kaur, 2019).These findings suggest that NLRP3 gene ablation or inhibition as an AD treatment target, though it is too early for clinical assessment.A genetic link between TREM2 and AD was established in 2013; thus, selective modulation of TREM2 may be another potential therapeutic strategy (Guerreiro et al., 2013).To date, several related agonist antibodies have been found to activate TREM2,including AL002 and AL002a/c (Cignarella et al., 2020; Wang et al., 2020b).However, novel TREM2-based treatments to manipulate microglial function currently face significant barriers to clinical application.Exciting achievements have been shown in stem cell-derived microglial replacement therapy in animal models.However, maintaining microglia in a beneficial state remains a challenge (Temple, 2023).Experimentally, bone marrow mesenchymal stem cell (MSC) transplantation can modulate microglial activation in APP/PS1 mice,showing both decreased inflammatory cells and increased anti-inflammatory cytokines, indicating alternative microglial activation.In addition, MSCs transplanted into APP/PS1 mice can ameliorate AD pathology and reverse spatial learning and memory declines (Zhang et al., 2016; Bagheri-Mohammadi, 2021).Although experimental and preclinical work suggests that stem cells are microglial regulators, with considerable therapeutic potential in AD, major safety and ethical issues remain to be overcome.
In summary, the effects of environmental factors on AD are often synergistic,multifaceted, and closely associated with individual genetic susceptibility.The compendium of research on the mechanisms by which environmental factors affect AD leave no doubt that microglia, the neuroimmunological brain cells,play an important role.Considering the increased incidence of patients with sporadic AD, who have genetic polymorphisms associated with microglia, it is important to understand the role of microglia in AD development, toward strategies targeting microglia for AD treatment.In the effort to intervene and control environmental AD risk factors at an early stage, an in-depth understanding of the role of microglia could be a key to AD treatment breakthroughs.
Author contributions:MZ wrote the manuscript and designed the figures.CL and XC reviewed the manuscript and gave valuable suggestions to this manuscript.LC and YC conceived the project and reviewed the manuscript.All authors approved the final manuscript.
Conflicts of interest:The authors declare that they have no competing interests.
Data availability statement:Not applicable.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
杂志排行

中国神经再生研究(英文版)的其它文章
- Glycolysis and glucose metabolism as a target for bioenergetic and neuronal protection in glaucoma
- MAP4K inhibition as a potential therapy for amyotrophic lateral sclerosis
- How do lateral septum projections to the ventral CA1 influence sociability?
- RNA sequencing of exosomes secreted by fibroblast and Schwann cells elucidates mechanisms underlying peripheral nerve regeneration
- Crosstalk among mitophagy, pyroptosis, ferroptosis,and necroptosis in central nervous system injuries
- Clustering of voltage-gated ion channels as an evolutionary trigger of myelin formation