亚低温干预通过改善线粒体功能抑制干扰素-α2b 诱导的人心肌细胞AC16 凋亡
2024-02-02王俊乾周灵杉朱友琦乙成成白明
王俊乾 周灵杉 朱友琦 乙成成 白明
目的:分析亚低温在I型干扰素(干扰素-α2b) 诱导的人心肌细胞AC16 凋亡过程中的作用及分子机制。
方法:不同浓度的干扰素-α2b 在不同时间点刺激人心肌细胞AC16,CCK-8 检测心肌细胞增殖;流式细胞仪检测干扰素-α2b 刺激心肌细胞后常温、亚低温对细胞的影响;线粒体Mito-Tracker 绿色荧光探针染色激光共聚焦成像观察线粒体形态的变化;流式细胞仪检测不同干预条件下线粒体膜电位变化情况;通过线粒体Mito-Tracker 绿色荧光探针染色激光共聚焦成像评估线粒体动力相关蛋白1(Drp1)与线粒体的共定位情况及不同干预条件对线粒体的影响;使用蛋白免疫印迹法检测不同干预条件下,磷酸化Drp1 Ser616、Drp1、核酶多聚二磷酸腺苷核糖聚合酶1(PARP1)、剪切型PARP1(cleaved-PARP1)蛋白表达变化。
结果:CCK-8 及流式细胞仪检测发现干扰素-α2b 抑制AC16 细胞增殖,并可诱导AC16 细胞凋亡,亚低温干预发挥心肌保护作用;不同干预条件下,AC16 细胞线粒体损伤程度不一, 表现为亚低温干预细胞具有较好的线粒体形态和更高的线粒体膜电位;Mito-Tracker 绿色荧光探针检测发现心肌细胞损伤时Drp1 从胞浆转移至线粒体中参与线粒体分裂,且亚低温干预发挥抑制作用;蛋白免疫印迹法检测发现亚低温干预后磷酸化Drp1 Ser616/Drp1 和cleaved-PARP1/PARP1 显著降低,且线粒体分裂抑制剂1(Mdivi-1)预处理可部分逆转上述现象。
结论:亚低温干预通过改善线粒体功能抑制I型干扰素-α2b 诱导的人心肌细胞AC16 凋亡。
心肌炎是以炎症浸润为主的心肌病变,并发症严重且预后较差[1]。柯萨奇病毒B3(CVB3)等病毒感染是心肌炎的主要原因[2]。病毒感染机体会诱导心肌细胞产生大量I 型干扰素,包括干扰素-α、干扰素-β 等多种类型,干扰素可激活免疫系统以限制病毒感染[3]。然而,异常和过量的I型干扰素产生也会导致心肌炎症损伤[4-5],目前临床治疗手段有限。线粒体稳态对于维持正常心肌生理功能至关重要,近年来,有研究发现,线粒体损伤参与了心肌炎的发病过程[6]。生理条件下,心肌细胞中线粒体不断融合和分裂以适应外部环境。当心肌细胞受到病毒、炎症因子、氧化应激因子、钙离子超载等因素刺激后,会引起线粒体损伤,受损的线粒体通过线粒体裂变发生断裂,最终依靠自噬实现自身降解[7]。线粒体裂变主要由线粒体动力相关蛋白1(Drp1)介导,Drp1 过表达可促使线粒体的分裂增多,导致线粒体裂变,促进了细胞凋亡[8]。Caspase-3 是细胞凋亡的关键执行蛋白。核酶多聚二磷酸腺苷核糖聚合酶1(PARP1)是与DNA 修复密切相关的一种酶,Caspase-3 主要剪切PARP1。剪切型PARP1(cleaved-PARP1)被认为是细胞凋亡的重要指标[9]。低温治疗可通过控制性降低患者核心温度以保护器官免受损伤影响,根据温度不同,有轻度低温(33℃~35℃)、中度低温(28℃~32℃)、深度低温(17℃~27℃)及超深度低温(<16℃)。在临床中,轻度低温又称为亚低温。有研究表明,亚低温治疗对心脏的保护作用由多种机制参与,包括代谢需求减少,心排血量需求减少,同时,亚低温能够减少活性氧释放,另外,可通过线粒体途径抑制细胞凋亡[10]。因此,我们推测亚低温可能抑制I型干扰素诱导的人心肌细胞AC16 凋亡,且这个过程与线粒体稳态调节有关。
1 材料与方法
1.1 试剂和仪器
实验时间为2022 年9 月至2022 年5 月。人心肌细胞AC16 购自宁波明舟生物科技有限公司。CCK-8、膜联蛋白V(Annexin V)-异硫氰酸荧光素(FITC)细胞凋亡检测试剂盒、线粒体绿色荧光探针(Mito-Tracker Green)试剂盒、羊抗兔IgG 购自上海碧云天生物技术有限公司。BCA 蛋白浓度测定试剂盒购自北京索莱宝科技有限公司,cleaved-PARP1兔单克隆抗体、PARP1 兔单克隆抗体、Drp1 兔单克隆抗体、磷酸化Drp1 Ser616 兔单克隆抗体、羊抗兔IgG 预吸附二抗购自英国Abcam 公司,ECL 化学发光剂购自北京普利莱基因技术有限公司。干扰素-α2b、线粒体分裂抑制剂1(Mdivi-1)均购自美国MCE 公司。
1.2 细胞培养与增殖检测
细胞培养:将人心肌细胞AC16 培养在含有10%胎牛血清和双抗的DMEM 培养基中,待细胞长到80%~90%时传代。细胞贴壁后用不同剂量干扰素-α2b(25、50、100、200、400 ng/ml)处理AC16 细胞4 h。
CCK-8 检测AC16 细胞活力:取对数生长期的AC16 细胞,在96 孔板中接种约5000 个细胞/孔,培养至细胞完全贴壁后按前述不同浓度的干扰素-α2b 处理细胞4 h,换液后继续培养24、48、72 h。向每孔加入10 μl CCK-8 反应液,孵育2 h 后用酶标仪测定450 nm 处的吸光度。
1.3 细胞分组和处理
将细胞放置于恒温培养箱培养,待细胞密度达70%~80%时分成7 组: 常温组、常温+干扰素-α2b组、常温+干扰素-α2b +Mdivi-1 组、亚低温组、亚低温+干扰素-α2b 组、亚低温+干扰素-α2b+ Mdivi-1 组。常温组为(37.0±0.5)℃,亚低温组为(33.0±0.5)℃。
常温条件下,常温+干扰素-α2b +Mdivi-1 组和亚低温+干扰素-α2b + Mdivi-1 组首先加入1µmol/L Mdivi-1 预处理AC16 细胞3 h,其余组正常培养。常温+干扰素-α2b 组、常温+干扰素-α2b+Mdivi-1 组、亚低温+干扰素-α2b 组、亚低温+干扰素-α2b + Mdivi-1 组细胞分别加入200 ng/ml干扰素-α2b,且细胞开始按照不同温度环境培养4 h;随即此四组细胞更换为常规培养液,继续在不同温度下培养48 h。常温组和亚低温组始终使用常规培养液,常温组始终常温培养,亚低温组则跟随亚低温+干扰素-α2b 组、亚低温+干扰素-α2b+ Mdivi-1 组的时间点进行亚低温培养。
1.4 流式细胞仪检测细胞凋亡
用0.25%胰酶[不含乙二胺四乙酸(EDTA)]消化的细胞,1 500 转/min 离心5 min 后弃去上清,收集细胞;用预冷磷酸缓冲盐溶液(PBS)重悬细胞2次,1500 转/min 离心5 min,洗涤细胞;按照说明书分别用Annexin V 和PI 染色,于避光条件下室温孵育10 min 后检测细胞凋亡。
1.5 Mito-Tracker Green 检测活细胞线粒体形态
按照说明书配制检测试剂,去除细胞培养液后,加入Mito-Tracker Green 染色工作液,置于37℃培养箱共孵育15~45 min。使用37℃预热的新鲜细胞培养液更换工作液后,用共聚焦显微镜观察。
1.6 流式细胞仪定量检测线粒体膜电位水平
以1×105个/孔密度将细胞接种于六孔板中,过夜后按上述分组处理细胞。胰酶消化后将细胞重悬于0.5 ml 细胞培养液中,加入0.5 ml JC-1 染色缓冲液混匀。置于37 ℃培养箱中孵育20 min 后,4 ℃条件下以600 g 转速离心3~4 min,取细胞沉淀。按照说明书配制适量JC-1 染色缓冲液(1×)后置于冰浴盒,使用预冷的JC-1 染色缓冲液(1×)洗涤沉淀2 次后重悬细胞,上机分析。
1.7 蛋白免疫印迹法(Western blot)检测磷酸化Drp1 Ser616、Drp1、cleaved-PARP、PARP 蛋白表达水平
制备样品并检测蛋白浓度,上样后跑胶。切胶后于65 V 电压转膜2 h。室温封闭1 h 后,加入对应一抗并置于4℃冰箱摇动过夜;室温孵育二抗1 h 后,显色并曝光,目的蛋白与内参蛋白的比值为相对蛋白含量。PARP1 抗体、cleaved-PARP1 抗体、Drp1 抗体、磷酸化Drp1 Ser616 抗体的稀释比例分别为1 : 1 000、1 : 5 000、1 : 1 000、1 : 1 000。
1.8 免疫细胞荧光评估Drp1 易位
细胞爬片后进行线粒体的荧光标记;使用3%甲醛溶液室温固定10~15 min,用PBS 洗涤3 次。将标本放置于12 孔板,用1% Triton-X 100 进行打孔穿透,室温孵育5~10 min。用PBS 洗涤3 次。在封口膜划分操作区域,每个样本位点加适量3%牛血清白蛋白(BSA)封闭液,室温封闭30 min。按1 : 250 的浓度稀释Drp1 抗体,置于湿盒4℃封闭过夜,PBS 洗涤3 次。按1 : 500 的浓度稀释山羊抗兔IgG 预吸附二抗和4’,6-二脒基-2-苯基吲哚二盐酸盐(Dapi)混合液,避光室温孵育30~60 min 后用PBS 洗涤3 次。封片固定后使用共聚焦显微镜观察拍照。
1.9 统计学方法
采用GraphPad 7.0 软件进行统计分析。所有数据以±s表示。组间比较采用单因素方差分析,多组间两两比较采用Tukey 方法。P<0.05 为差异有统计学意义。
2 结果
2.1 常温组和常温+干扰素-α2b 组的心肌细胞活力的比较
24 h 时,常温组与常温+干扰素-α2b 组(干扰素-α2b 剂量25、50、100、200、400 ng/ml)的心肌细胞活力的差异均无统计学意义。48 h、72 h 时,常温组与常温+干扰素-α2b 组(干扰素-α2b 剂量25 ng/ml)的心肌细胞活力差异无统计学意义。从干扰素-α2b 剂量50 ng/ml 开始,随着干扰素-α2b剂量(100、200、400 ng/ml)的增加,常温+干扰素-α2b 组在48 h、72 h 时的细胞活力均低于常温组(P均<0.05)。
2.2 心肌细胞凋亡的比较(图1)

图1 流式细胞仪检测细胞凋亡
与常温组相比,常温+干扰素-α2b 组、亚低温+干扰素-α2b 组细胞凋亡率升高,亚低温组细胞凋亡率降低,差异均有统计学意义(P均<0.01);与常温+干扰素-α2b 组相比,亚低温组、亚低温+干扰素-α2b 组的细胞凋亡率明显降低,差异均有统计学意义(P均<0.01);与亚低温组相比,亚低温+干扰素-α2b 组细胞凋亡率明显升高,差异有统计学意义(P<0.01)。由此可见,亚低温能够抑制干扰素-α2b 诱导的细胞凋亡。
2.3 心肌细胞线粒体形态的比较(图2)
常温组细胞线粒体呈梭形;常温+干扰素-α2b组细胞线粒体肿胀;常温+干扰素-α2b + Mdivi-1组细胞线粒体肿胀变形得到缓解;亚低温组细胞线粒体细长,其面积和周长指数更大,圆形度显著降低;亚低温+干扰素-α2b 组细胞线粒体肿胀程度减轻;亚低温+干扰素-α2b + Mdivi-1 组细胞线粒体大部分正常。
2.4 心肌细胞线粒体膜电位水平(图3)
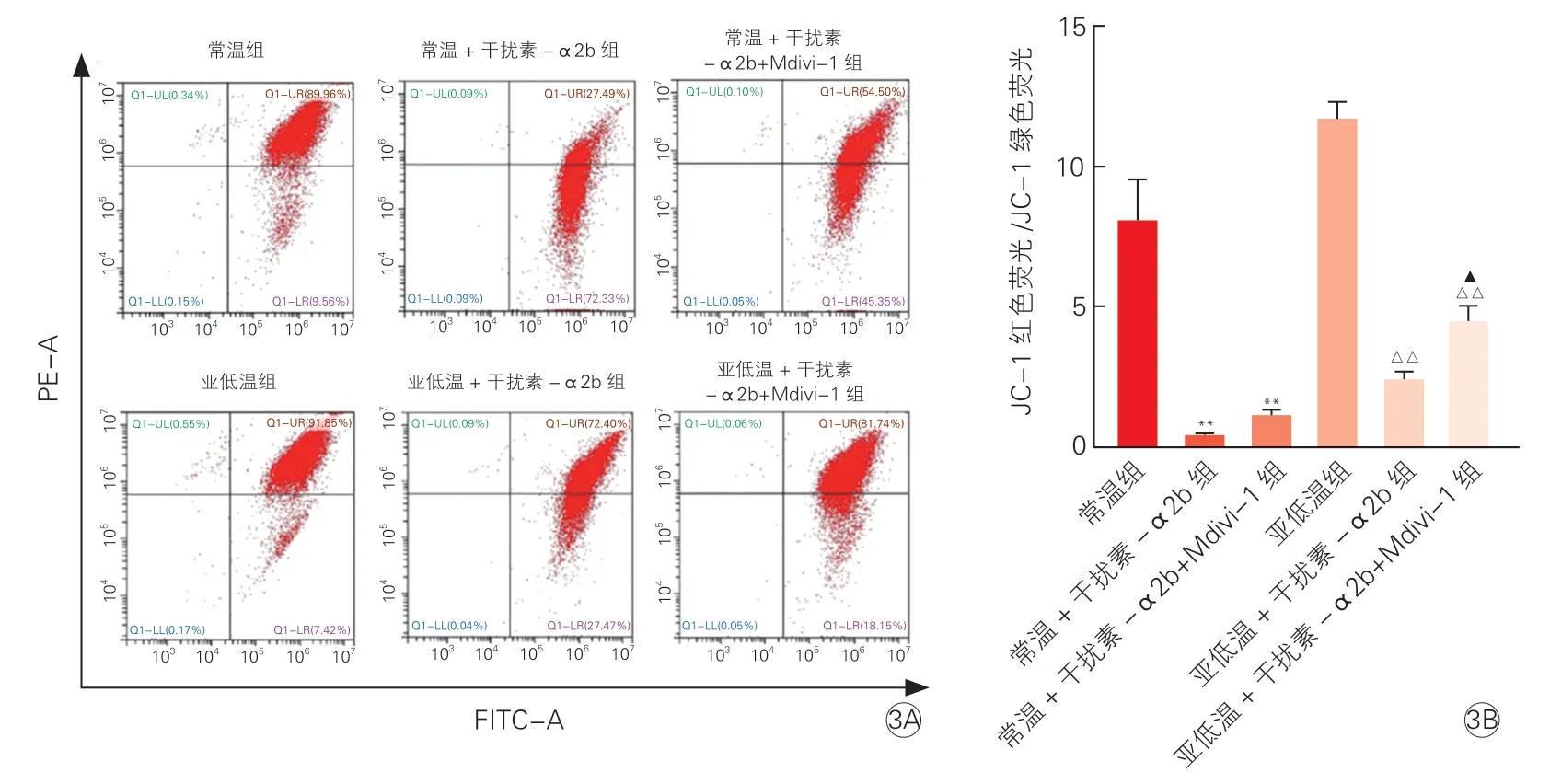
图3 流式细胞仪检测线粒体膜电位水平
与常温组相比,常温+干扰素-α2b 组及常温+干扰素-α2b+Mdivi 组细胞线粒体膜电位红色荧光强度明显减弱,绿色荧光强度均明显增强,线粒体膜电位均明显降低(P均<0.01)。与亚低温组相比,亚低温+干扰素-α2b 组、亚低温+干扰素-α2b+Mdivi组线粒体膜电位均明显降低(P均<0.01)。与亚低温+干扰素-α2b 组相比,亚低温+干扰素-α2b+Mdivi组细胞线粒体膜电位红色荧光强度明显增强,绿色荧光强度明显减弱,线粒体膜电位明显升高(P<0.05)。这些结果表明,亚低温及Mdivi-1 干预能够上调干扰素-α2b 诱导的线粒体膜电位。
2.5 cleaved-PARP1/PARP1、 磷酸化Drp1 Ser616/Drp1 的蛋白表达水平(图4)
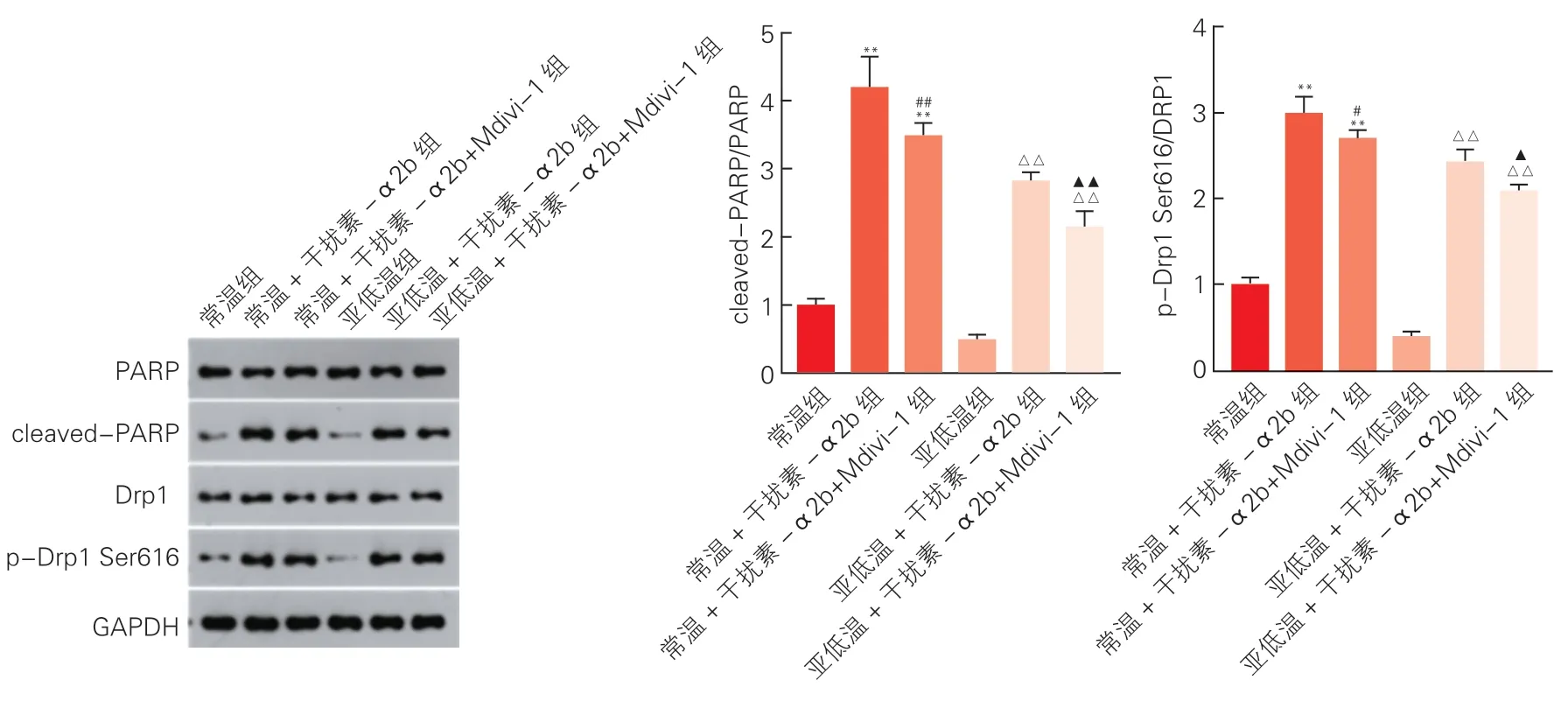
图4 蛋白免疫印迹法检测心肌细胞cleaved-PARP1/PARP1 以及磷酸化Drp1 Ser616/Drp1 的蛋白表达水平
与常温组相比,常温+干扰素-α2b 组、常温+干扰素-α2b+Mdivi 组磷酸化Drp1 Ser616/Drp1、cleaved-PARP1/PARP1 蛋白表达水平均明显上调(P均<0.01)。与常温+干扰素-α2b 组比较,常温+干扰素-α2b+Mdivi 组磷酸化Drp1 Ser616/Drp1、cleaved-PARP1/PARP1 蛋白表达水平明显下调(P<0.05)。与亚低温组相比,亚低温+干扰素-α2b 组、亚低温+IFN-α2b+Mdivi 组磷酸化Drp1 Ser616/Drp1、cleaved-PARP1/PARP1 蛋白表达水平均明显上调(P均<0.01)。与亚低温+干扰素-α2b 组比较,亚低温+干扰素-α2b+Mdivi 组磷酸化Drp1 Ser616/Drp1、cleaved-PARP1/PARP1 蛋白表达水平明显下调(P<0.01)。
2.6 免疫细胞荧光评估心肌细胞中Drp1 易位情况(图5)

图5 免疫细胞荧光评估心肌细胞中Drp1 易位情况
常温组细胞质中Drp1 荧光弱,细胞线粒体几乎未募集到Drp1;常温+干扰素-α2b 组细胞Drp1荧光最强,Drp1 向线粒体易位最强,表明在常温状态下干扰素对心肌细胞具有更强的损伤作用;常温+干扰素-α2b+Mdivi-1 组细胞Drp1 荧光较常温+干扰素-α2b 组减弱,向线粒体的迁移受限制;亚低温组Drp1 荧光最弱,细胞线粒体未观察到Drp1易位;亚低温+干扰素-α2b 组细胞Drp1 荧光线粒体募集的Drp1 较少;亚低温+ 干扰素-α2b+Mdivi-1组细胞线粒体募集的Drp1 进一步减少。
3 讨论
病毒可以触发免疫反应产生I型干扰素,对限制病毒感染至关重要,但异常和过量的I型干扰素产生会导致心肌炎症损伤[4]。干扰素-α 诱导的心肌细胞是心肌炎体外模型的常用方法[5]。本文探讨了亚低温对I型干扰素(干扰素-α2b)诱导的人心肌细胞AC16 中线粒体相关功能的调控作用和机制,发现亚低温干预能够明显抑制干扰素-α2b 诱导的心肌细胞凋亡,且与显著增加的线粒体膜电位水平相关。先前的研究表明,治疗性低温32℃~34℃可能对新生儿缺氧缺血性脑病或心脏骤停患者的神经系统发挥保护作用[11-12]。也有研究表明,亚低温能够降低细胞的钙负荷,维持心肌细胞能量代谢和调节组织水平的葡萄糖,也可清除活性氧[13]。另外,亚低温可增强心肌细胞的小分子泛素相关修饰物(small ubiquitin-related modifier,SUMO)化修饰(SUMOylation),使心肌细胞中SUMO1 与靶蛋白结合的能力显著提高,从而抑制凋亡相关蛋白的表达发挥拮抗心肌凋亡的作用[14]。亚低温对心肌细胞的保护作用在本研究中也达到证实。
近年来,一些研究认为线粒体损伤参与了心肌炎的发病机制[6,15]。在生理条件下,线粒体融合和裂变通常保持动态平衡,以适应细胞环境[16]。而在病理状态下,病毒感染、高糖、高脂、炎症、氧化应激、钙超载等多种危险因素都可能导致线粒体损伤。受损的线粒体通过线粒体裂变分离并通过自噬降解,而健康部分通过融合重新形成新的线粒体。线粒体动力学的不平衡会导致线粒体形态异常伸长或断裂,最终导致细胞凋亡[17]。线粒体裂变主要Drp1 介导,该蛋白在激活时易位到线粒体外膜,通过翻译后修饰,活性受到严格调控,包括磷酸化、棕榈酰化、苏甲酰化、S-亚硝基化和泛素化[8]。在细胞发生凋亡前,Drp1 从胞质溶胶到线粒体的易位主要受两个关键位点的磷酸化调节:一个导致丝氨酸 616 的活化,另一个导致丝氨酸 637 的失活[18]。Drp1 从胞质转移到线粒体,导致线粒体通透性转换孔(MPTP)打开,通过这些孔道,线粒体内细胞色素C (Cyt-C)等促凋亡因子释放后与凋亡蛋白酶活化因子1(APAF-1) 结合激活细胞凋亡蛋白酶-9(Caspase-9)。活化后的Caspase-9 可以使下游Caspase-3 酶原活化转变为Caspase-3。它是激活凋亡的主要效应物,作用于不同的靶分子,最终导致细胞分解和程序性细胞死亡[19]。我们的研究发现, 干扰素-α2b 诱导的人心肌细胞AC16 Drp1蛋白ser616 位点表达水平升高,表明干扰素-α2b促进心肌线粒体分裂,而亚低温干预部分延缓了了这一病理生理学过程。另一方面,本研究检测干扰素-α2b 诱导的人心肌细胞AC16 中cleaved-PARP1与总PARP1 的表达占比,观察到cleaved-PARP1 占比增高,表明与干扰素-α2b 诱导的细胞凋亡有关,同时上述过程可能与DNA 损伤有关。
亚低温治疗能够减弱 Cyt-C 的释放并且可以降低Caspase-9 的表达从而抑制线粒体诱导的细胞凋亡[20-21]。本研究发现,亚低温干预能明显减轻干扰素-α2b 导致的线粒体损伤,包括改善线粒体的形态,以及降低线粒体的凋亡率。对于线粒体膜电位,亚低温干预能增加膜电位水平。另外,目前许多研究提示抑制 Drpl 激活以及抑制 Drp1 向线粒体膜上转移可以抑制过度的线粒体分裂从而减轻缺血再灌注损伤后的细胞凋亡。而亚低温对 Drp1 活性的影响仍少见报道,本研究发现,亚低温能够降低Drp1 蛋白的表达水平,减少Drp1 向线粒体易位。Mdivi-1是Drp1 的关键抑制剂。既往研究示,Mdivi-1 预处理可通过抑制Drp1 表达上调,减轻心肌细胞死亡[22]。此外,Mdivi-1 能够通过抑制线粒体膜电位耗散、改善线粒体ATP 能量产生、减轻ROS 过度产生和保护氧化应激诱导的细胞损伤,改善线粒体功能障碍[23]。本研究发现,Mdivi-1 可降低Drp1 蛋白的表达水平,减少Drp1 向线粒体易位,并使线粒体膜电位恢复。
综上所述,亚低温干预通过改善线粒体功能抑制干扰素-α2b 诱导的人心肌细胞AC16 凋亡,表明治疗性低温有望成为病毒性心肌炎治疗的有效手段。
利益冲突:所有作者均声明不存在利益冲突
