基于土壤宏基因组的聚磷酸激酶的筛选与鉴定
2024-02-02章素平高森浩李王馨月张锦豪杨诗韵尤忠毓
章素平,高森浩,李王馨月,张锦豪,杨诗韵,尤忠毓*
1(嘉兴南湖学院 新材料工程学院,浙江 嘉兴,314001)2(嘉兴大学 生物与化学工程学院,浙江 嘉兴,314001)
腺嘌呤核苷三磷酸(adenosine triphosphate,ATP)是一种含有2个高能磷酸键的化合物,是生物体各类生命活动所需能量的重要来源[1]。在生物催化生产高价值的产品时,ATP也是最常见的能量供体,但ATP本身价格昂贵,经济上不允许在反应过程中直接添加ATP[1]。目前最常见的ATP供应方式是ATP再生系统,即将一个合成ATP的酶催化反应与消耗ATP的酶催化反应进行偶联,使其能够以廉价的原料实现ATP的循环再生[2]。截止目前,ATP再生系统所使用的酶主要是激酶,包括聚磷酸激酶、乙酸激酶、腺苷酸激酶、氨基甲酸激酶、丙酮酸激酶等,其中聚磷酸激酶的应用较为广泛[1-2]。
聚磷酸激酶(polyphosphate kinase,PPK,EC 2.7.4.1)能够以多聚磷酸盐(polyPn)为磷酸供体催化ADP生成ATP,其反应过程为:ADP+polyPn→ATP+polyPn-1,其中polyPn可分为短链polyPn(n<10)和长链polyPn(n>10),由于短链polyPn的合成简单、价格低廉,因此,基于PPK和短链polyPn的ATP再生系统能够得到有效的应用[3-4]。目前来源于大肠杆菌(Escherichiacoli)、嗜热栖热菌(Thermusthermophilus)、嗜热聚球藻(Thermosynechococcuselongatus)、白色链霉菌(Streptomycesalbulus)等多种生物的PPK已被鉴定并用于ATP再生系统的构建,实现了5-氨基酮戊酸、D-木酮糖-5-磷酸、谷胱甘肽、ε-聚赖氨酸等多种化合物的酶法合成[3,5-7]。不同来源的PPK具有不同的催化特性与底物偏好性(不同链长的polyPn),适用于不同的ATP依赖型酶催化反应,因此,筛选并开发新型的PPK将有利于扩大ATP再生系统的应用范围。
宏基因组是指生境中所有微小生物遗传物质的总和,是新型生物催化剂的重要来源[8]。基于宏基因组的生物催化剂筛选避免了微生物的分离纯化过程,加快了新型生物催化剂的开发进程。近年来,利用宏基因组技术已经发现了大量的生物催化剂,例如,双功能纤维素酶、木聚糖酶、酯酶、内切-β-1,4-葡聚糖酶等[9-12]。本研究以土壤宏基因组为研究对象,利用直接PCR扩增法筛选新型聚磷酸激酶基因,并实现其在大肠杆菌中的重组表达,为新型ATP再生系统的开发奠定基础。
1 材料与方法
1.1 材料
1.1.1 实验试剂
土壤基因组DNA快速抽提试剂盒、DNA-EZ Reagents L土壤腐植酸清除剂、SanPrep柱式质粒DNA小量抽提试剂盒、SanPrep柱式DNA胶回收试剂盒、T载体PCR产物克隆试剂盒、Taq Plus DNA聚合酶、限制性内切酶(NcoI和XhoI)、ATP、ADP、多聚磷酸钠(polyP6)等均购自生工生物工程(上海)股份有限公司。
1.1.2 菌株、质粒及培养基
E.coliDH5α、E.coliBL21(DE3)、pET28a均由本实验室保藏,分别用于基因的克隆及表达。LB培养基(g/L):胰蛋白胨10,酵母提取物5,氯化钠10,固体培养基加入20 g/L琼脂,必要时加入相应抗生素:氨苄青霉素(100 μg/mL)或卡那霉素(50 μg/mL)。
1.2 实验方法
1.2.1 土壤采集
土壤样品采自浙江省内农田、果园、湿地公园等,取样深度距土壤表面10~15 cm。
1.2.2 土壤宏基因组DNA的提取
利用土壤腐植酸清除剂除去各土壤样品中的腐植酸,再利用土壤基因组DNA快速抽提试剂盒提取土壤样品的总DNA,利用1%琼脂糖凝胶电泳检测提取样品。
1.2.3 聚磷酸激酶基因的获取
从GenBank数据库中下载来源于E.coli(EcPPK,WP_115187122)、T.thermophilus(TtPPK,BCZ86902)、T.elongatus(TePPK,QLL30020)、Neisseriameningitidis(NmPPK,AAA85674)、Alcaligenesfaecalis(AfPPK,KGP01591)的PPK氨基酸序列,通过多重序列比对搜索保守序列,根据保守氨基酸序列设计简并引物。以土壤宏基因组DNA为模板,进行PCR扩增,并将扩增产物与T-载体连接后转化至E.coliDH5α感受态细胞中,挑取单克隆送上海生工测序。将测序结果输入GenBank数据库中进行比对,选择序列一致性较高且定义为PPK的序列为研究对象。根据数据库中对应的全长序列,直接设计特异性引物,以相应的土壤宏基因组DNA为模板,通过PCR获得全长编码基因,通过与T-载体连接后测序验证。
1.2.4 聚磷酸激酶基因的序列分析
使用MEGA5对潜在PPK蛋白序列和已知的PPK序列进行多重比较,构建系统发育树(建树方法:Neighbor-joining,Bootstrap method检测1 000次,建树模型:Poisson model)。利用ProtParam在线工具(https://web.expasy.org/protparam/)对PPK的理化性质进行分析;利用ProtScale(https://web.expasy.org/protscale/)对疏水性进行分析;利用TMHMM Server v.2.0(https://services.healthtech.dtu.dk/services/TMHMM-2.0/)分析跨膜结构;利用SignalP 5.0(http://www.cbs.dtu.dk/services/SignalP/)分析信号肽;利用SWISS-MODEL在线服务(http://www.swissmodel.expasy.org/)进行同源建模,构建三级结构模型[13],用PDBsum Generate在线服务(http://www.ebi.ac.uk/thornton-srv/databases/pdbsum/ Generate.html)对3D结构进行评估,蛋白质结构通过PyMOL展示。
1.2.5 聚磷酸激酶的重组表达及分离纯化
根据序列分析的结果,重新设计表达引物,并在引物中引入NcoI和XhoI酶切位点。将重新PCR扩增的PPK基因与表达载体pET28a连接,转化至E.coliDH5α感受态细胞中,利用菌落PCR筛选阳性克隆,并通过酶切及测序验证重组质粒构建的正确性。
将测序验证正确的表达质粒转化至表达宿主,重组菌株接种至含50 μg/mL卡那霉素的LB培养基中,37 ℃培养至OD600值等于0.6,再向培养基中加入终浓度为0.1 mmol/L的异丙基-β-D-硫代半乳糖苷(isopropyl-β-D-thiogalactopyranoside,IPTG),15 ℃条件下诱导20 h,4 ℃、8 000 r/min离心10 min收集菌体,并重悬于100 mmol/L PBS(pH 7.0)缓冲液中,超声破碎细胞,12 000 r/min离心收集上清和沉淀,利用SDS-PAGE检测蛋白的表达情况。
利用Ni-TED 6F色谱柱和AKTA pure层析系统分离目的蛋白。首先,用平衡缓冲液(50 mmol/L、pH 7.0的磷酸盐缓冲液+300 mmol/L NaCl)平衡色谱柱,上样后用洗脱液1(平衡缓冲液+50 mmol/L咪唑)对非特异性吸附的杂蛋白进行洗脱,再用洗脱液2(平衡缓冲液+300 mmol/L咪唑)洗脱目的蛋白,通过SDS-PAGE检测蛋白的分离情况。
1.2.6 重组聚磷酸激酶的酶活检测
以ADP和多聚磷酸钠为底物,检测聚磷酸激酶的活力,反应体系(10 mL)如下:50 mmol/L pH 7.5磷酸盐缓冲液,5 mmol/L ADP,10 mmol/L多聚磷酸钠,10 mmol/L MgCl2,加入1 mL酶液,40 ℃反应300 min,沸水浴5 min终止反应,12 000 r/min离心后取上清液,利用HPLC检测ATP的生成量。HPLC检测条件:色谱柱为C18柱;流动相:V(磷酸盐缓冲液)∶V(甲醇)=90∶10;检测波长254 nm;流速为1 mL/min;柱温35 ℃。
1.2.7 重组聚磷酸激酶的应用
灵菌红素缩合酶PigC可以在ATP存在下催化合成灵菌红素及其类似物[14]。本试验将重组聚磷酸激酶与灵菌红素缩合酶PigC联用,合成灵菌红素类似物。反应体系(1 mL)如下:50 mmol/L pH 7.5磷酸盐缓冲液,1 mmol/L ADP,5 mmol/L多聚磷酸钠,5 mmol/L MgCl2,0.2 mmol/L 4-甲氧基-2,2-二吡咯-5-羧甲基乙醛(MBC),0.2 mmol/L 2,4-二甲基-3-乙基吡咯,聚磷酸激酶和灵菌红素缩合酶PigC粗酶液各100 μL,25 ℃水浴反应1 h,加入酸性丙酮终止反应。
2 结果与分析
2.1 土壤宏基因组DNA的提取
土壤样品中常存在大量的腐殖酸,会影响DNA的提取及后续的分子生物学操作。为了获得高质量的宏基因组DNA,本实验首先利用腐植酸清除剂提前处理土壤样品,再利用土壤基因组DNA快速抽提试剂盒提取土壤总DNA。经1%琼脂糖凝胶电泳检测,结果如图1所示,各泳道样品均显示单一高分子量条带,且无明显的拖尾、弥散等现象,说明所提取的DNA相对完整,无降解,质量较高,有利于后续实验的开展。
2.2 聚磷酸激酶基因的克隆
从GenBank中获取5条不同来源的PPK氨基酸序列,利用Clustalw进行多重序列比对,根据比对结果确定了2处保守性较高的序列(图2-A,图2-B),两个保守序列相距330个氨基酸左右。以保守的氨基酸序列为基础,设计了一对简并引物:P1:5′-ARTARCAAYYTRGAYGARTTYTWY-3′,P2:5′-YTCRTCRAA-YCKNGCCHTYMMYTC-3′。以土壤宏基因组DNA为模板,进行PCR扩增。扩增产物经1%琼脂糖凝胶电泳检测,结果显示(图2-C),不同的模板扩增后得到多种大小不一的片段,包括250、350、550、750、1 000、2 000 bp等。根据简并引物设计的距离,选择大小为1 000 bp的片段进行胶回收后与T-载体连接,转化至E.coliDH5α感受态细胞中,经蓝白斑筛选,挑取阳性克隆送上海生工测序。
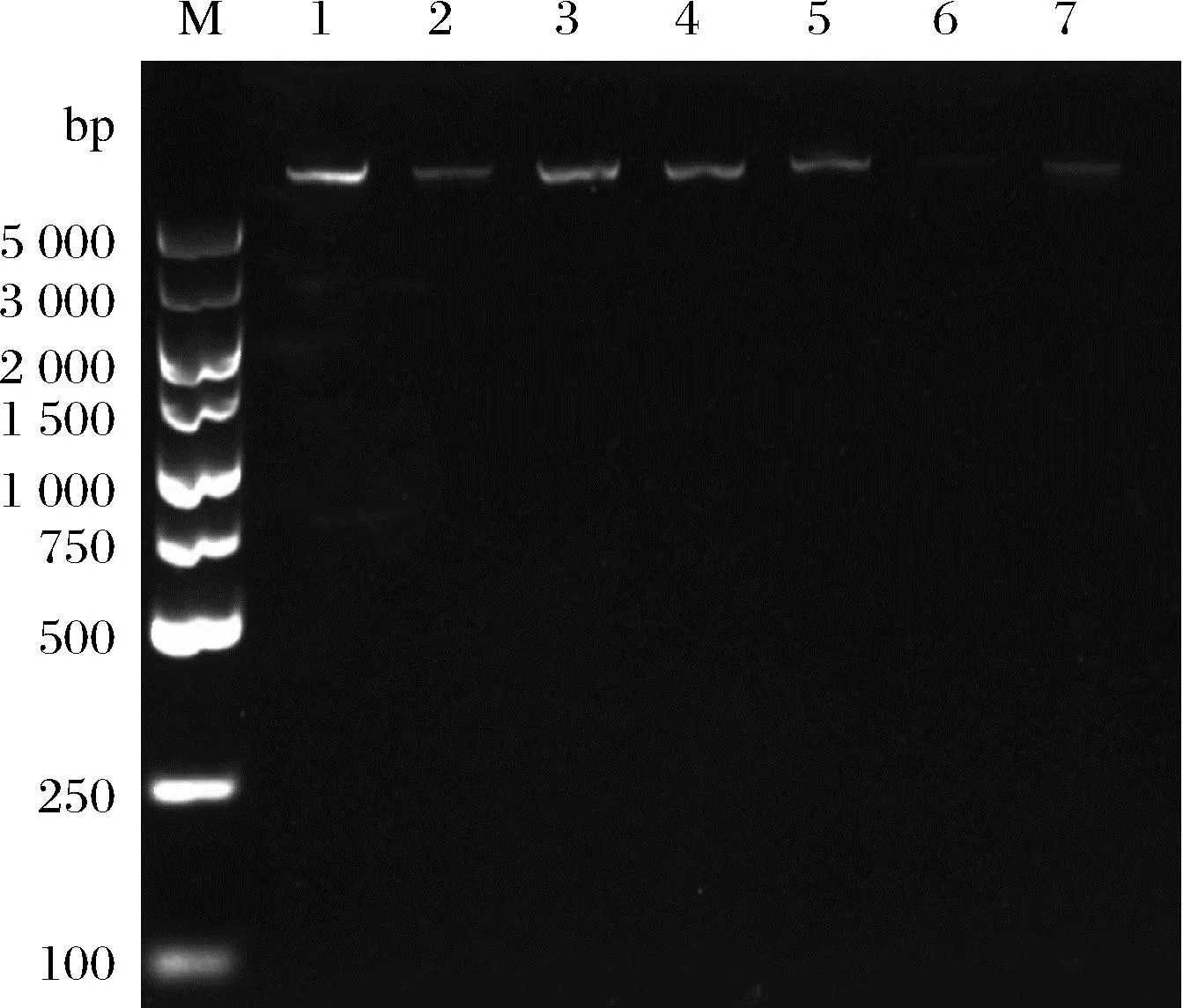
M-DNA Marker;1-7-不同土壤样品的基因组DNA图1 土壤宏基因组DNA电泳图Fig.1 Agarose gel electrophoresis analysis of soil metagenomic DNA
测序结果显示,共获得2条不同的DNA序列,将其编码的氨基酸序列在GenBank中进行BlastP分析,结果显示,其中一条序列与E.coli的PPK序列一致性达99%,另一条序列与Serratiamarcescens的PPK序列一致性达98.9%。查阅文献可知,来源于E.coli的PPK已被广泛研究[15],而关于S.marcescens的PPK研究鲜见报道。因此,选择S.marcescens的PPK为后续研究对象,以GenBank中S.marcescens的PPK基因全长序列直接设计引物(F:5′-ATGGGTCAGGAAAAGCTCTACATCG-3′,R:5′-CTACTGTCCTGGTTGTTCCAGAGC-3′),以相应土壤宏基因组DNA为模板,通过PCR扩增全长,琼脂糖凝胶电泳显示,扩增产物为2 000 bp左右的片段(图2-D),经T-A克隆后测序,结果显示,在所克隆的片断中包含一个2 064 bp的开放阅读框,编码一个由687个氨基酸残基组成的蛋白质,序列分析表明,该蛋白即为S.marcescens的PPK(SmPPK)。
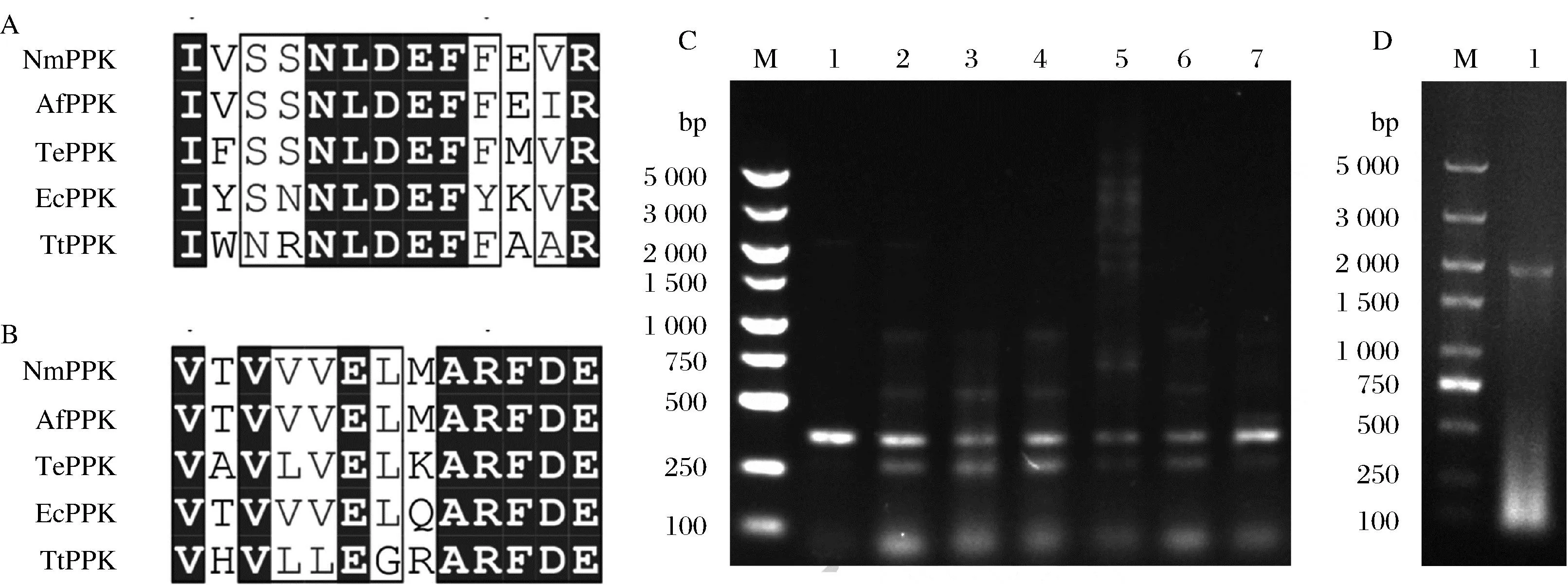
A-上游保守氨基酸序列;B-下游保守氨基酸序列;C-不同宏基因组DNA为模板的PCR产物;D-候选基因的全长PCR产物图2 聚磷酸激酶基因的筛选Fig.2 Screening of polyphosphate kinase genes
2.3 SmPPK的生物信息学分析
从GenBank中筛选11条来源于不同微生物的PPK序列,利用MEGA5构建系统发育树,确定不同来源PPK之间的进化距离。结果显示(图3-A),不同来源的PPK聚到2个大的分支上,其中SmPPK与Serratianevei、Gibbsiellaquercinecans等来源的PPKs在同一分支上,说明它们的亲缘关系较近。根据文献报道[1],PPK可以分为PPK1和PPK2两大家族,由系统发育树可知SmPPK属于PPK1家族。多重序列比对显示(图3-B),SmPPK与来源于E.coli、T.thermophilus、T.elongatus、N.meningitidis、A.faecalis的PPK氨基酸序列一致性分别为87.9%、23.4%、30.1%、31.5%、31.0%。
利用ProtParam在线工具对SmPPK的理化性质进行了分析。SmPPK由687个氨基酸残基组成,其中亮氨酸(Leu)占比最高为11.5%,半胱氨酸(Cys)占比最低为0.3%,该蛋白的分子式为C3599H5696N996O1023S17,分子质量79.8 kDa,等电点8.8。SmPPK分子中带正电荷氨基酸残基(Arg+Lys)为96个,带负电荷氨基酸残基(Asp+Glu)为90个。利用ProtScale对SmPPK的亲/疏水性进行分析,结果显示(图3-C),SmPPK的第584位的Val疏水性最强(分值为1.689),第190、191位的Arg亲水性最强(分值为-2.656),结合ProtParam分析可知,SmPPK的总平均亲水指数为-0.294,因此,SmPPK为亲水性蛋白。利用TMHMM Server v.2.0和SignalP 5.0对SmPPK的跨膜结构和信号肽进行分析。结果表明(图3-D、图3-E),SmPPK没有跨膜区和信号肽。
2.4 SmPPK的同源建模
为了解SmPPK的空间结构,以E.coli来源的PPK晶体结构(PDB ID:1XDP)为模板,在SWISS-MODEL中进行同源建模,结果显示(图4-A),SmPPK的空间结构呈典型的L型结构,包含20个α螺旋和27个β折叠。利用PDBsum Generate对所构建的模型进行评估,拉氏图(Ramachandran plot)显示(图4-B),该模型中处于最佳区域的残基比例为88.1%,处于允许区域的残基比例为11.70%,处于不允许区域的残基仅占0.2%,说明该模型较合理。对比E.coli的PPK可知[15],SmPPK可分为4个结构域(图4-C):N-端结构域(residues 1~106)、“头部”结构域(residues 107~321)和2个C-端结构域(residues 322~500,501~687),由N-端结构域和2个C-端结构域构成了一个底物进出的通道。ZHU等[15]报道了EcPPK的活性中心由His435、Asp470、His592和Glu623组成,根据序列比对发现,SmPPK中与上述4个氨基酸残基相对应的分别是His433、Asp468、His590和Glu621,而且以上4个氨基酸残基在其他PPK中高度保守(图3-B),因此,推测His433、Asp468、His590和Glu621构成了SmPPK的活性中心(图4-D)。
2.5 SmPPK的重组表达
根据序列分析结果,设计表达引物:P3:5′-CATGCCATGGGTCAGGAAAAGCTCTAC-3′,P4:5′-ATAC TCGAGCTGTCCTGGTTGTTCCAGAGC-3′,下划线部分为NcoI和XhoI酶切位点。通过PCR扩增目的基因SmPPK,与表达载体pET28a经NcoI和XhoI双酶切后连接,构建重组表达载体pET28a-SmPPK,并转化至克隆宿主。重组质粒经单/双酶切验证(图5-A),双酶切产物可见约2 000 bp的目的基因和5 400 bp的载体,单酶切产物可见大于5 400 bp的单一条带,说明重组表达载体构建成功,进一步通过测序验证序列的正确性。
将测序验证正确的重组质粒pET28a-SmPPK转化至表达宿主E.coliBL21(DE3),经0.1 mmol/L IPTG诱导后,进行SDS-PAGE电泳检测,结果显示(图5-B),在80 kDa的位置有特异性表达条带,与目的蛋白理论值一致。细胞破碎上清和沉淀中均有目的条带,说明SmPPK在表达过程中形成了包涵体,可以通过降低诱导温度、减少诱导剂浓度等方法,增加其可溶性表达。SmPPK的C末端携带His标签,因此,采用镍离子亲和层析的方法对细胞破碎液上清中的目的蛋白进行分离纯化,SDS-PAGE电泳检测显示(图5-C),利用含50 mmol/L咪唑的洗脱液去除杂蛋白,再用含300 mmol/L咪唑的缓冲液洗脱目的蛋白,可以得到电泳纯的SmPPK。
2.6 SmPPK的活性检测及应用
为验证SmPPK在ATP合成中的效率,本试验以ADP和多聚磷酸钠为底物,考察了SmPPK合成ATP的时间进程,结果显示(图6-A),在反应初期的60 min内,ATP的产率快速升高,后期随着时间的延长,ATP产率增速逐渐变缓,当反应240 min时,ATP产率达到最大值46.5%。为进一步验证SmPPK在ATP再生系统中的应用潜能,以SmPPK和灵菌红素缩合酶PigC为催化剂构建了双酶偶联催化合成灵菌红素类似物的反应体系,结果显示(图6-B),当反应体系中只有PigC时,反应液的颜色较浅,即灵菌红素类似物的产量较少;当体系中同时存在PigC和SmPPK时,反应液的颜色明显变深,即灵菌红素类似物产量增加。以上结果表明,SmPPK具有较好的ATP合成能力,可用于构建ATP再生系统并应用于ATP依赖的酶催化反应。
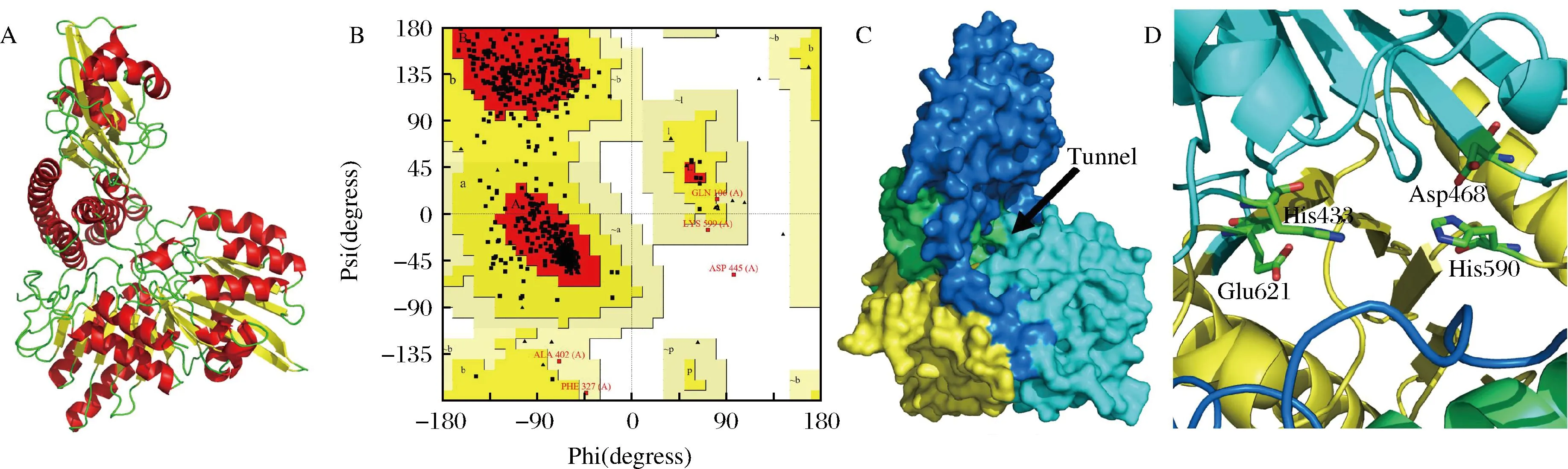
A-SmPPK的3D结构模型;B-SmPPK结构模型拉氏图;C-SmPPK的结构域模型(N-端结构域为绿色;“头部”结构域为蓝色; C端结构域分别为青色和黄色);D-SmPPK的活性位点(His433、Asp468、His590和Gln621)图4 SmPPK的3D结构预测Fig.4 Predictive 3D structure model of SmPPK
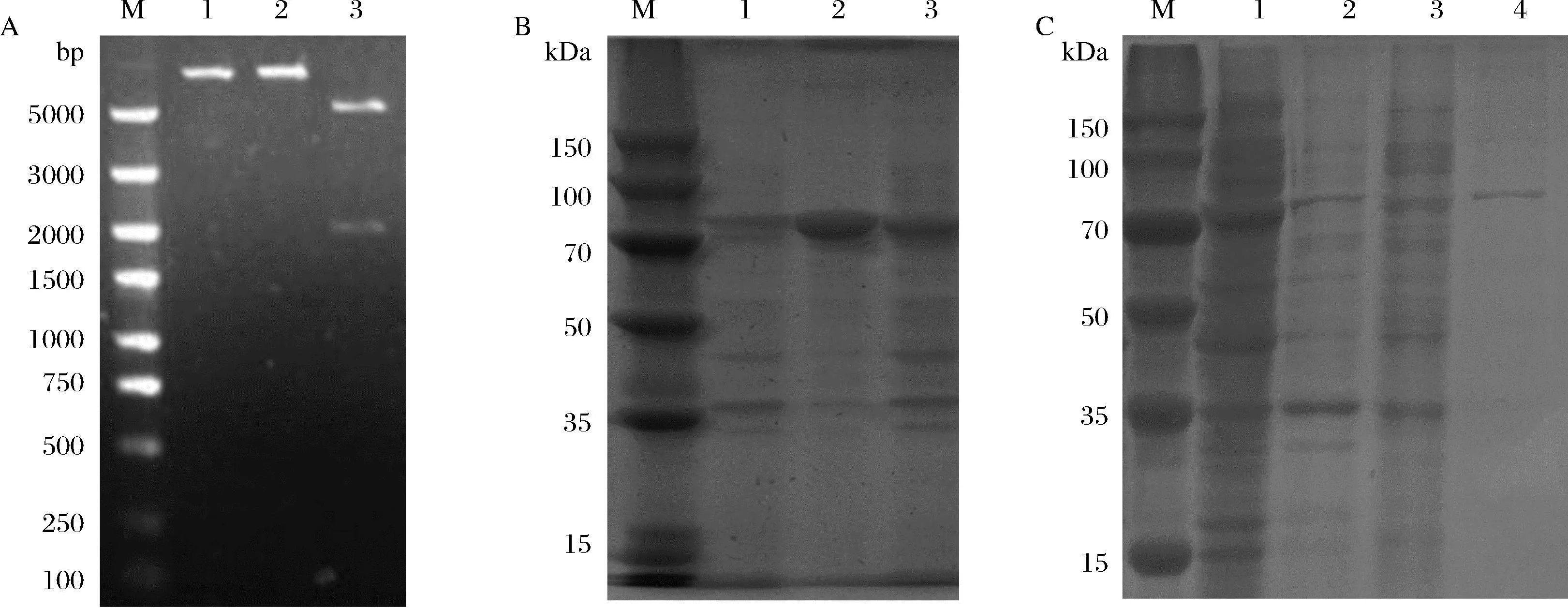
A-重组质粒pET28a-SmPPK的酶切验证(M-DNA Marker;1-pET28a-SmPPK的Nco I酶切产物;2-pET28a-SmPPK的Xho I酶切产物; 3-pET28a-SmPPK双酶切产物);B-SmPPK诱导表达的SDS-PAGE分析(M-Protein Marker;1-未诱导细胞的裂解物;2-诱导细胞的裂解沉淀;3-诱 导细胞的裂解上清);C-SmPPK的纯化(M-Protein Marker;1-裂解上清;2-穿透峰;3-50 mmol/L咪唑洗脱的样品;4-300 mmol/L咪唑洗脱的样品)图5 SmPPK的重组表达Fig.5 Recombinant expression of SmPPK
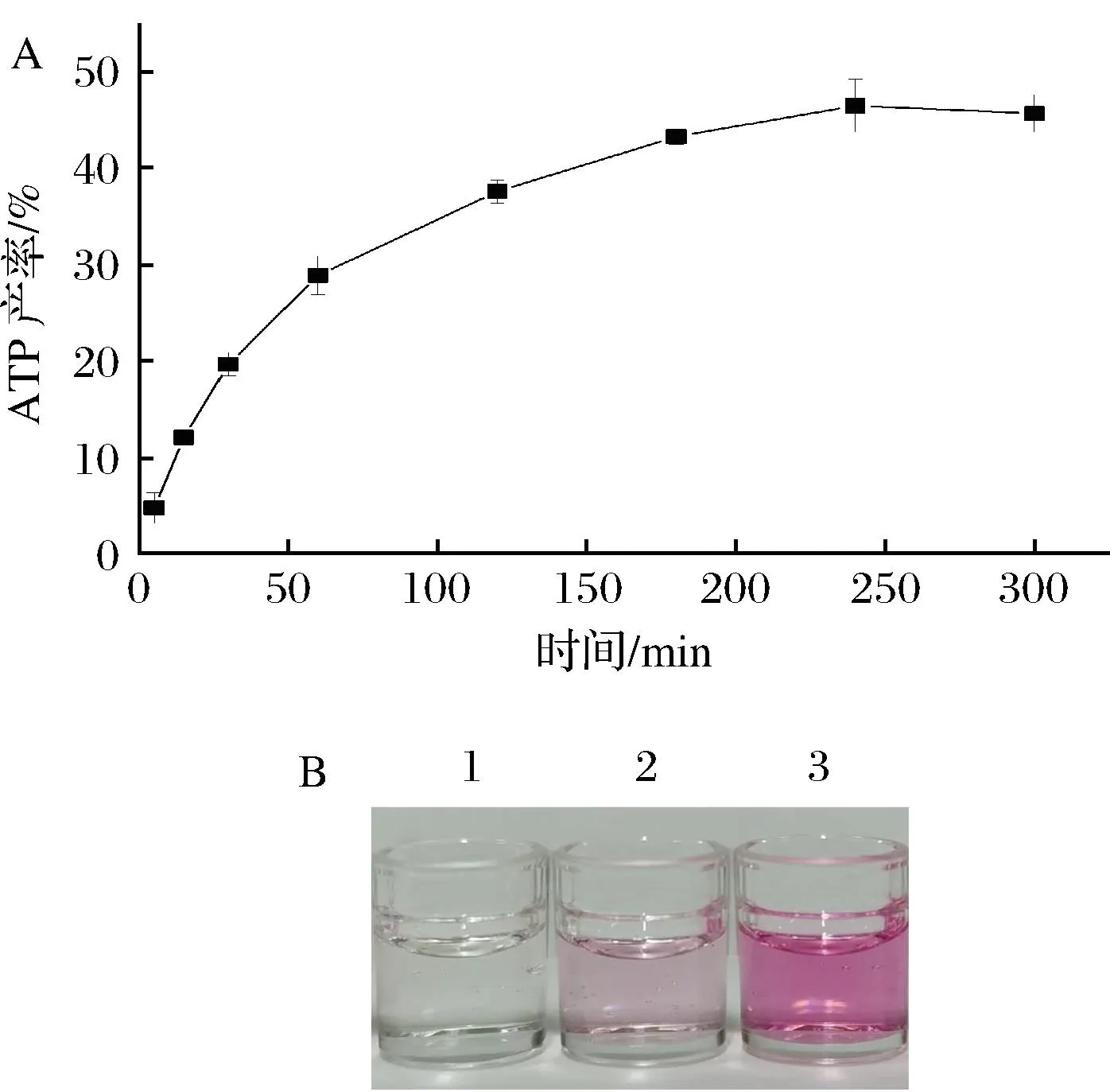
A-SmPPK催化合成ATP的时间进程;B-基于SmPPK的ATP 再生系统在灵菌红素类似物合成中的应用 (1-空白对照;2-含PigC但不含SmPPK;3-含PigC和SmPPK)图6 SmPPK的活性检测及应用Fig.6 Enzyme activity and application of SmPPK
3 结论与讨论
生物催化过程因具有效率高、选择性高、环境友好等优点,广泛应用于高附加值化学品的生产[16]。ATP作为一种重要的辅因子,参与许多需要能量的生物催化过程,但直接向反应体系中添加ATP不仅增加了生产成本,而且会产生多种副产物影响后续的产品分离[7,17]。因此,构建廉价、高效的ATP再生系统对于ATP依赖的生物催化过程显得至关重要。聚磷酸激酶因其底物廉价易得,已成为ATP再生系统构建的首选酶之一。截止目前,已报道的成功应用于ATP再生的聚磷酸激酶数量有限[1],因此,挖掘新型聚磷酸激酶构建ATP再生系统将有利于推动ATP依赖的生物催化过程的应用进程。
宏基因组技术避开了微生物纯培养的过程,扩大了可研究的微生物范围,是一种筛选新酶基因的有效方法[18]。本研究基于聚磷酸激酶的保守序列设计简并引物,从土壤宏基因组中筛选到一条来源于S.marcescens的聚磷酸激酶基因SmPPK,序列分析表明,SmPPK与E.coliPPK具有较高的序列一致性,同属于PPK1家族。该家族PPK通常由600~700个氨基酸组成,对ATP具有较高的底物偏好性,部分家族成员属于膜结合蛋白[2,6],但SmPPK经TMHMM分析表明不具有跨膜区。同源建模显示,SmPPK由4个结构域组成典型的L型空间结构,其活性中心主要由His433、Asp468、His590和Glu621组成[15],催化过程可能通过酶自身的磷酸化实现,而His433和His590在磷酸化过程中发挥重要作用[19]。
本研究采用大肠杆菌系统实现了SmPPK的重组表达,并利用镍离子亲和层析对重组蛋白进行了分离纯化,获得了电泳纯的重组SmPPK。酶活检测显示,SmPPK可以在多聚磷酸钠的存在下,实现ATP的合成,其最高产率为46.5%,该产率高于源自E.coliMG1655的PPK(15.5%)和源自Rhodobactersphaeroides的PPK(20.1%),但低于源于泗阳鞘氨醇杆菌的PPK(71%)[4]。在利用SmPPK构建ATP再生系统与灵菌红素缩合酶PigC耦合时,成功实现灵菌红素类似物的合成。因此,SmPPK在ATP再生领域具有较好的研究和应用价值。本研究下一步将对SmPPK的酶学特性及催化机理进行更加深入的研究,以了解该酶适用的催化条件,提高其合成ATP的产率,为更多ATP依赖的生物催化过程提供ATP再生系统。
