Agn-1Si(n=5-10)团簇结构与性质研究
2024-02-01邹松霖罗有华
邹松霖, 罗有华
(华东理工大学 物理学院, 上海200237)
1 引 言
纳米团簇是由几个到几千个原子或分子通过一些物理或者化学结合力组成的相对稳定的微观或亚微观聚集体[1]. 它是一种联结微观和宏观物质结构的新物质形态. 团簇研究是多学科交叉领域,是基于物理和化学的交叉点,成长于材料科学. 与此同时,计算机和计算机技术的迅速发展,对团簇的结构性质研究可以从第一性原理出发,进行从头算和动力学模拟. 团簇物理学是一门发展迅速的新型交叉性科学,涉及的范围非常丰富和广泛[2-4].
Ag团簇作为贵金属[5]的一种在最近几十年来因为其在传感[6]、催化[7]、生物制药[8]等方面的广泛应用,已经受到各界高度关注并展开各种理论和实验上的研究[9],比如Jin等人[10]提出了纯银团簇(n= 4-16)的基态结构并发现比较稳定的Ag6,Ag8和Ag14团簇. 其中关于非幻数14价电子结构也通过类晶体场效应[11]进行了解释. 这种几何扭转将超原子分子轨道分裂,随着团簇价电子的变化,整个团簇也发生从球形到狭长形再到扁圆形的变化[12]. 同时也出现了很多将其他元素掺杂在Ag团簇中[13,14],为微观团簇的研究提供了新的思路. Si在纳米尺度上有很多卓越的性质[15],所以很难预见将Si原子放在一个纯银团簇中会有什么样的反应. Kiran和Bernd[16]通过研究Si原子掺入Aun-团簇和Agn-团簇(n= 2-56和5-82)中的光电子图谱得出Si原子可以改变团簇中自由电子数,在Au团簇中更多是提供电子,在Ag团簇中既可以获得电子也可以提供电子,可以提供或者失去最多四个电子. 在Ag54Si-团簇已经开始和Ag55团簇拥有十分相似的几何结构了. Junais和Udo[17]系统研究Agn和Agn-1Si(n= 5-12)的中性和阴阳离子团簇的结构和光学性质,并发现在Si原子掺杂下,主要在银团簇外部与Ag原子形成共价键,Agn团簇光学性质变化明显,使吸收峰展宽并且衰减. Zhao等人[18]的研究发现,纯银团簇掺入Si原子,Si原子的位置更多是团簇的表面,在低能量的异构体中,有更多的Si-Ag键的团簇更加的稳定.
本文主要利用无差别结构搜索软件和第一性原理优化软件,系统性的寻找了Agn-1Si团簇(n= 5-10)的低能结构. 近年研究的工作主要针对银团簇,对Si掺杂银团簇的研究还较少,其中的团簇结构主要通过手工或者遗传算法等方法得到,可能会错过能量更低的结构. 在确定每个尺寸的最低能量结构后,具体分析其稳定性增加的原因,为银团簇参加非金属元素的实验和理论研究提供了新的依据.
2 研究方法
本文采用基于第一性原理出发的密度泛函理论对Agn-1Si(n=5-10)团簇结构和性质进行计算研究. 首先使用Crystal structure AnaLYsis by Particle Swarm Optimization(CALYPSO)[18,20,21]来获得团簇的初始构形. CALYPSO基于粒子群算法,包含产生限制性结构,键特征矩阵消除相似结构,加入随机结构增加结构多样性一般以80%作为保留上一代结构的比例和一些其他的技术. 一般结构能在十代左右收敛,每一代计算30个结构. 通过CALYPSO得到的大量初始结构,再使用基于平面波密度泛函理论计算程序VASP进行优化,使用的赝势文件分别是PAW_PBE_ Ag和PAW_PBE_ Si.平面波基矢截断能为300 eV,每个团簇放在15×15×15 Å3的晶格中,在这个足够大的空间,相邻的团簇之间的相互作用可以忽略不计,电子能量的收敛条件为1×10-8eV. 在布里渊区域上集成时只考虑Γ点(k= 0). 没有任何对称或自旋约束的情况下,结构被弛豫,直到原子间力小于0.02 eV/Å.
3 研究结果与讨论
3.1 Agn团簇和Agn-1Si团簇(n = 5-10)结构
通过CALYPSO找出低能结构体,利用VASP进行更高精度的优化后找出能量最低的结构. 如图一上半部分为Agn团簇(n= 5-10)结构,与文献[10]所得一致,Ag5团簇具有C2v对称性,呈平面梯形排列. Ag6团簇具有D3h对称性,是在Ag5的基础上生长为正三角结构,这样的结构将分子轨道进行了扁平化扭曲,团簇分子轨道1P劈裂为两部分,导致团簇外价电子按照1s21p4排布[12],形成稳定的满壳层结构. 从Ag7团簇开始,团簇从二维平面结构向三维立体结构转变,Ag7具有D5h对称性. Ag8团簇具有Td对称性,开始形成双层结构,上下平面分别四个Ag原子构成,两个平面长对角线相互垂直. Ag9团簇具有Cs对称性,在双层结构的底部填充一个Ag原子,另一平面的一个Ag原子被挤出平面,填充到团簇的顶部,形成封闭的空心几何结构. Ag10团簇具有D2d对称性,在增加一个Ag原子后,依然保持封闭的空心结构. 可以看出明显的生长特征,随着团簇尺寸的增加,Ag团簇从平面结构转变为扁平面结构,之后趋向更加致密的准空心球状.
Agn-1Si团簇结构如图一下半部分所示,其中浅色小球代表Ag原子,深色小球代表Si原子. Ag4Si团簇具有C4v对称性,Si原子戴帽于四个Ag形成的平面之上. Ag5Si团簇具有C1对称性,是在Ag4Si团簇基础上添加一个Ag原子,所添Ag原子处于Si原子斜上方,底部四个Ag原子组成的正方形片面略微变化为准平面. 比文献[17]中八面体结构低0.29 eV. Ag6Si团簇具有Cs对称性,再添加的Ag原子在远离Si原子的另一侧,位于Ag原子平面下方,同时四个Ag原子又重新围成一个正方形平面,这时开始Si原子以及表现出位于团簇外侧的特点. Ag7Si团簇没有随着Ag原子的加入将Si笼罩,而是在远离Si原子的区域重新构成Ag准平面. 比文献[17]二帽八面体结构低0.11 eV. Ag8Si团簇是在Ag7Si团簇基础上,在远离Si原子的区域上添加一个Ag原子,可以看出远离Si原子的位置已经开始重新形成银平面. Ag9Si团簇具有Cs对称性,Si原子在团簇外侧. 所得能量最低的Agn-1Si(n= 5-10)团簇结构也有明显的生长特征,Si原子戴帽于银团簇外侧,增加的Ag原子在远离Si原子的地方继续形成准平面结构.
3.2 Agn团簇和Agn-1Si团簇(n=5-10)电子性质比较
团簇电子性质是否稳定主要通过HOMO-LUMO 能隙的大小来判断. 图二(a-c)分别代表了团簇HOMO-LUMO能隙,平均结合能和二阶能量差,方点代表Agn团簇,圆点代表Agn-1Si团簇. 通过图二(a)可以看出,Agn团簇中HOMO-LUMO 能隙在0.25到2.28 eV之间,最大值是Ag8团簇,Ag6团簇相对小一点. Agn-1Si团簇中HOMO-LUMO 能隙在0.31-2.44 eV之间,Ag4Si团簇最大,Ag6Si团簇和Ag8Si团簇相对小一点. 两种团簇都表现出奇偶震荡的特点,主要与单电子不能形成闭合电子结构有关. Ag8和Ag4Si团簇外层都有8个价电子,它们HOMO-LUMO的大能隙的原因是团簇1P和1D分子轨道之间的能量相差较大.
3.3 Agn团簇和Agn-1Si团簇(n = 5-10)平均结合能和二阶能量差
团簇的稳定性可以通过平均结合能Eb来验证,平均结合能越大,说明团簇越稳定,平均结合能Eb的定义为:
Eb(Agn)=[nE(Ag)-E(Agn)]/n
(1)
Eb(Agn-1Si)=[(n-1)E(Ag)+
E(Si)-E(Agn-1Si)]/n
(2)
通过图二(b)可以看出,Si原子的加入使银团簇更加稳定,在Agn团簇中,随着团簇尺寸的增加,团簇的稳定性有一个增加的趋势,特别在奇数团簇到偶数团簇会有明显的增加. 由于偶数银团簇没有多余单电子,可以形成电子对. 在小尺寸范围(n< 8),团簇中Ag原子的个数比是否拥有成对电子更能影响平均结合能. 在Agn-1Si团簇中,并没有随着团簇尺寸增加而更加稳定的趋势,相反是出现了明显的奇偶震荡现象,拥有偶数价电子的团簇更加的稳定. 团簇结构也是趋向准平面化,没有出现一个更加紧凑的结构. Ag4Si团簇平均结合能最高,Ag6Si和Ag8Si团簇相对较低,说明满足幻数结构的团簇能量结构也更稳定.
团簇之间的相对稳定性可以通过二阶能量差(Δ2E)来验证,二阶能量差越大,说明该团簇附近尺寸的稳定性更高. 二阶能量差的定义为:
Δ2E(Agn)=E(Agn-1)+E(Agn+1)-2E(Agn)
(3)
Δ2E(Agn-1Si)=E(Agn-2Si)+
E(Agn)-2E(Agn-1Si)
(4)
通过图二(c)可以看出,方点代表的Agn团簇和圆点代表的Agn-1Si团簇整体呈现出奇偶震荡的特征,主要原因是未成对电子导致,在Ag8处为最大值,说明Ag8在n=5-10尺寸的团簇中相对稳定性最高,Ag6团簇、Ag10团簇次之. 在Agn-1Si团簇中Ag4Si相对稳定性最高,Ag6Si团簇和Ag8Si团簇次之. 综合以上HOMO-LUMO 能隙,平均结合能Eb,二阶能量差Δ2E可以得出,在Agn团簇和Agn-1Si团簇中,团簇尺寸在5-10之间,Ag8团簇和Ag4Si团簇,都是满足价电子为八的幻数结构,在三个方面都表现出稳定性最高.
3.4 差分电荷密度分析
差分电荷密度图可以反映原子成键过程后电荷的转移,图三为Agn-1Si团簇(n= 5-10)的差分电荷图,图中深色部分代表电荷密度增加,浅色部分代表电荷密度减少. 通过图可以看出,Ag4Si和Ag5Si团簇中,电荷集中在Si原子和各个Ag原子之间. Ag6Si团簇中,与Si原子相邻的五个Ag原子之间电荷增加,剩余的一个Ag原子与相邻的两个Ag原子之间电荷增加. 在Ag7Si团簇中,与Si原子相邻的五个Ag原子之间电荷增加,另外两个Ag原子与两个靠近Si原子的Ag原子之间,电荷略微增加,说明这连个Ag原子之间有金属相互作用. Ag8Si团簇与Ag9Si团簇中也是在与Si原子相邻的Ag原子之间电荷增加较多,而远离Si原子的Ag原子附近电荷转移较少,主要是与附近的Ag原子形成金属键. 总的来说,Si原子的加入,使Si原子与Ag原子之间产生了比较强烈的共价相互作用,加上Ag原子与Ag原子之间的金属相互作用,提升了团簇的稳定性.
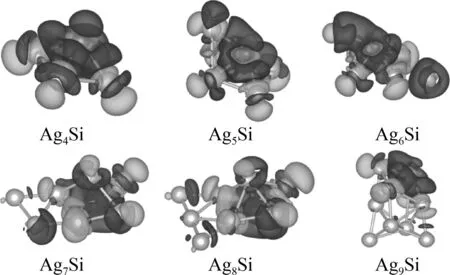
图3 Agn-1Si团簇(n=5-10)差分电荷密度图. Fig. 3 The deformation charge densities of Agn-1Si clusters (n=5-10).
4 结 论
本文采用密度泛函理论对Agn-1Si团簇(n= 5-10)进行了研究. 首先通过粒子群优化算法(CALYPSO)对Agn和Agn-1Si团簇进行无差别搜索,纯Ag团簇与之前的研究吻合,得到的含硅团簇Ag5Si,Ag7Si结构比之前文献具有更低的能量. 将Agn和Agn-1Si团簇从HOMO-LUMO 能隙分析其电子性质的变化,平均结合能比较团簇稳定性以及二阶差分能比较团簇的相对稳定性. 经过分析得出,Ag8与Ag4Si团簇具有较高的稳定性,其团簇价电子都满足八电子幻数结构. 之后通过差分电荷密度分析了Agn-1Si团簇(n= 5-10)电荷密度转移. 可以发现由于Si原子的加入,整个团簇结构变得更加紧致,比如相同团簇尺寸的纯Ag团簇还表现出平面或者亚平面的结构特征,Agn-1Si团簇已经是完整的三维结构. 在Si原子的加入后,与Si原子相邻的Ag原子向Si原子聚集,产生强烈的共价相互作用. 但是随着团簇尺寸的增加,并没有出现Ag原子将Si原子笼罩的现象,与Si原子较远的Ag原子还是趋于平面结构,它们也主要是靠Ag原子之间相互的金属键连接. 说明Si原子的加入可以有限的增强整个结构的稳定性,但在Si原子作用范围之外的区域,整个团簇还是靠Ag原子之间的金属键构成平面或亚平面结构.
