TiVNbTa 难熔高熵合金的吸放氢动力学
2024-01-25张李敬杨继荣
龙 雁,张李敬,杨继荣,王 芬*
(1 广东省金属新材料制备与成形重点实验室,广州 510640;2 华南理工大学 机械与汽车工程学院,广州 510640)
氢作为一种清洁且高效的能源获得了人们的广泛关注,常见的储氢方法有高压气态储存、低温液态储存、低温压缩储存和固态储存[1-2]。其中固态储存是通过固体储氢材料以化学或物理方式储存氢气,具有储氢密度高、安全性好的优点,被视为储氢最可行的方案之一[3]。
高熵合金 (high-entropy alloys,HEAs),又称为多主元高熵合金。Yeh 教授最初基于成分,定义由5 种或更多(通常小于13 种)主要元素组成,每种元素含量居于5%~35%(原子分数,下同)之间,并且可以包含一定微量元素(通常小于5%)的合金为高熵合金[4]。随着高熵合金研究的进一步发展,有研究者认为构型熵大于1.36R(R为气体常数)的四主元合金也可以称为高熵合金[5-7]。高熵合金具有的“四大核心”效应使得这类合金具有与传统合金不同的组织和特性[8-10]。严重的“晶格畸变效应”导致晶格应变的增加,从而有利于间隙位置容纳更多的氢原子[11-12],使高熵合金在储氢领域拥有巨大的应用潜力[13-14]。
Montero 等[15]在TiVZrNb 难熔高熵合金中加入Mg 元素制备新型储氢合金,合金在室温下短时间内即可吸氢,吸氢量达到2.7%(质量分数),经过5 次吸放氢循环后吸氢量稳定在2.4%(质量分数)。Mg 元素的添加使得合金储氢循环性能得到改善。Zlotea等[16]使用Ta 元素替换TiVZrNbHf 难熔高熵合金中的V 元素,研究Ta 元素对TiZrNbHfTa 难熔高熵合金的晶体结构、晶格畸变和吸氢性能的影响。研究表明,使用Ta 元素替换V 元素后使得合金晶格畸变加剧,吸氢量得到提升(氢原子与金属原子比为H/M=2.5)。Shen 等[17]研究TiZrHfMoNb 高熵合金的相变特性和储氢性能。结果表明,TiZrHfMoNb 高熵合金吸氢后由单相BCC 结构转变为FCC 结构,合金的氢化物在302 ℃左右开始放氢,脱氢后合金恢复单相BCC 结构,具备可逆的吸氢反应。现阶段对于高熵合金储氢性能的研究主要聚焦于不同成分的高熵合金储氢量或循环性能方面,而对于高熵合金吸放氢行为及其动力学机制等方面的认识还非常有限,因此对高熵合金的吸放氢动力学研究是有必要的。
本工作通过真空电磁感应悬浮熔炼技术制备TiVNbTa 合金样品,随后对TiVNbTa 难熔高熵合金进行吸(放)氢性能测试,以研究合金粉末的吸(放)氢动力学,探究该合金的吸(放)氢动力学机制,并获得TiVNbTa 难熔高熵合金的吸(放)氢表观活化能,为高熵合金吸(放)氢动力学研究提供参考。
1 实验材料与方法
使用真空电磁感应悬浮熔炼技术对等原子比的Ti,V,Nb,Ta 纯金属颗粒进行熔炼,金属颗粒的纯度均大于99.9%(质量分数),反复熔炼10 炉次使合金铸锭成分均匀化,制备出TiVNbTa 合金铸锭。采用线切割将合金铸锭切成1 cm×1 cm×4 cm 的长方体,并用砂纸打磨干净用于吸氢活化处理,活化后块体合金粉化成较细的粉末,随后使用该氢化粉末进行脱氢处理,并用于吸(放)氢动力学研究。
采用HORIBA-LA960S 型激光粒度分析仪对氢化粉末进行粒度分析,取大于2 g 的粉末置于无水乙醇分散介质中进行测试,粉末颗粒激光折射率近似为2.2。采用NOVA-NANO-SEM 430 型扫描电子显微镜对氢化粉末的形貌进行分析。采用PCI-MULTI TRACK 型多通道储氢性能测试仪对TiVNbTa 难熔高熵合金进行吸(放)氢动力学测试。采用X’ pert Powder 多位自动进样X射线衍射仪(CuKα,λ=0.154 nm)对吸氢前后、脱氢后的合金粉末进行XRD 测试,以确定其相组成。采用STA449-F3 型DSC 同步热分析仪对吸氢前、后的高熵合金进行热分析,以确定合金在升温过程中发生的还原反应或相变。使用铑坩埚为测试容器,测试温度为303~1673 K(以10 K/min 升温速率),在测试过程中通入氩气进行保护。
2 结果与分析
2.1 氢化合金粉末的形貌和相组成
铸态合金经多次重熔后,得到单相固溶体组织,但存在一定的枝晶偏析。成分分析结果表明,铸态合金中各合金元素的含量分别为:Ti 24.5%,V 25.0%,Nb 24.7%,Ta 25.8%,合金的实际成分与名义成分较为接近。铸态合金首次吸氢活化后出现粉化现象,图1(a)为粉化后的氢化粉末表面形貌图,粉末呈不规则的颗粒状。图1(b)为氢化合金粉末的粒度分布图,其平均颗粒尺寸为114 μm,在后续的吸-放氢过程中,合金粉末的尺寸及形貌并未发生明显改变。
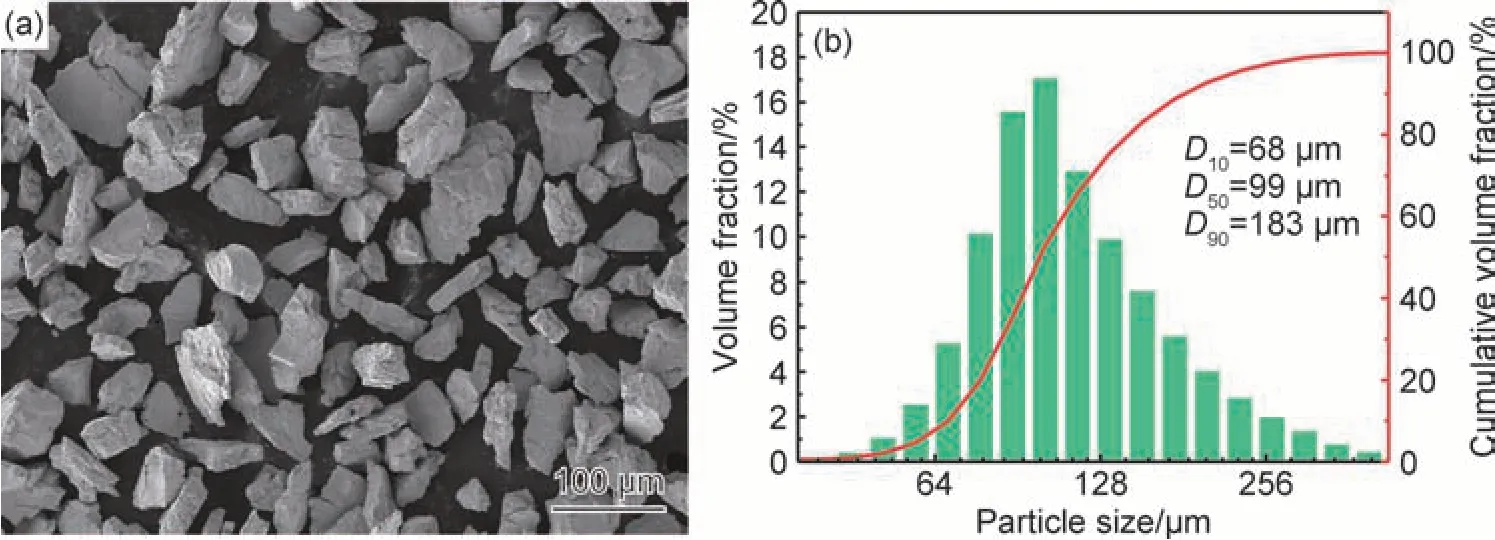
图1 TiVNbTa 块体合金氢化后形成的粉末形貌(a)及粒度分布(b)Fig.1 Powder morphology(a) and particle size distribution(b) of TiVNbTa bulk alloy after hydrogenation
对TiVNbTa 难熔高熵合金吸氢前合金铸锭、吸氢后的氢化粉末和放氢后的脱氢粉末试样进行XRD测试,其结果如图2 所示。吸氢前试样为单相BCC 结构,吸氢后试样发生相应的吸氢转变从而生成了新相。根据PDF 卡片校对鉴定,初步判定生成的新相分别为TiH1.971,Nb0.696V0.304H 和Nb0.498V0.502H2,空间群分别为和合金脱氢后恢复单相BCC 结构,这表明TiVNbTa 难熔高熵合金的吸氢反应为可逆反应。
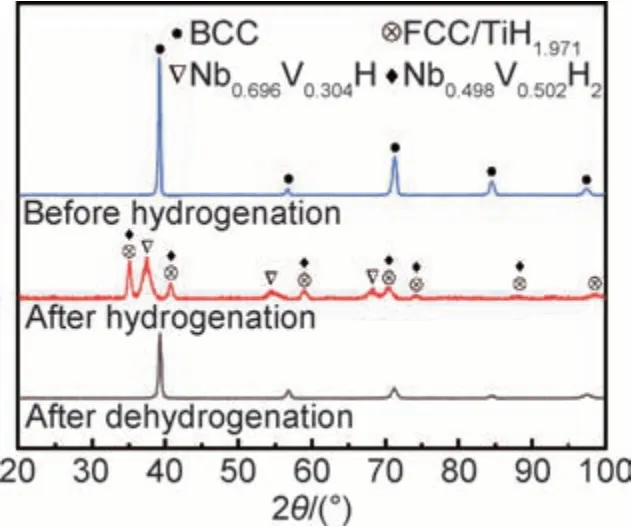
图2 TiVNbTa 难熔高熵合金吸氢前后、放氢后对应的XRD 图谱Fig.2 XRD patterns corresponding to TiVNbTa refractory high-entropy alloy before, after hydrogenation and after dehydrogenation
图3 为TiVNbTa 难熔高熵合金吸氢前后的DSC热分析曲线,可以看到未吸氢合金在升温过程中没有吸热峰和放热峰出现,表明吸氢前的合金在升温过程中并未出现相变。通过图3 中吸氢后的 TiVNbTa 难熔高熵合金粉末试样的DSC 热分析曲线,可以看到氢化合金粉末受热后存在3 个明显的吸热峰。在吸(放)氢反应过程中,吸氢反应为放热反应,而放氢反应为吸热反应,因此,吸氢后DSC 曲线中3 个明显的吸热峰对应着3 个放氢反应,表明氢化物发生分解需要进行三步反应,对应的吸热温度为放氢温度,分别为519,593 K 和640 K。三步放氢反应可能与3 种不同的氢化产物相对应。由于放氢动力学曲线为恒温测试,而DSC 曲线为变温测试,测试的温度在一定速率下升高。而合金的放氢过程需要一定时间,DSC 测试的氢化物分解温度存在一定的滞后性,因此合金吸氢后的DSC 曲线中氢化物分解的起始温度比实际值偏高。同时该DSC 曲线在1228 K 还存在一个放热峰,主要的原因可能为测试温度过高导致试样发生氧化。
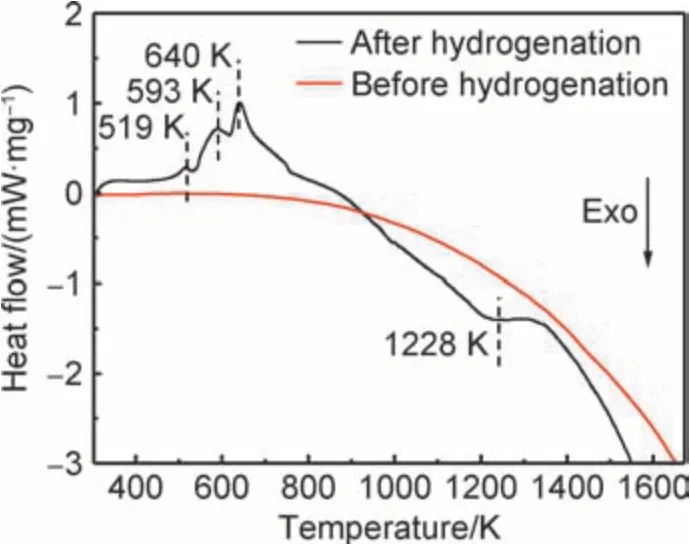
图3 吸氢前后高熵合金的DSC 曲线Fig.3 DSC curves of high-entropy alloy before and after hydrogen absorption
2.2 吸氢动力学
在723 K,2 MPa 条件下对TiVNbTa 高熵合金粉末进行1 h 的高温活化,图4(a)为活化后的难熔高熵合金在不同温度条件下的恒温吸氢动力学曲线,初始压力为1.8 MPa。TiVNbTa 难熔高熵合金粉末试样在423,573 K 和723 K 温度下吸氢均不存在孕育期,活化后便迅速吸氢,试样吸氢量经过约30 s 便可达到平衡吸氢量的90%,分别在约280,500 s 和900 s 后可达平衡吸氢量,平衡吸氢量(质量分数)分别为0.96%,0.90% 和0.77%。TiVNbTa 合金的平衡吸氢量随着温度的升高而降低。从图4(b)中可以看到,试样平衡氢压随着温度的升高而增加,符合气体状态方程。
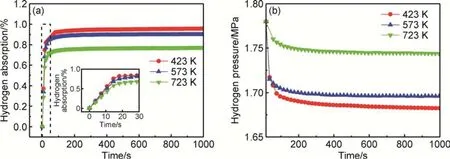
图4 TiVNbTa 难熔高熵合金吸氢曲线(a)吸氢动力学曲线;(b)吸氢压力曲线Fig.4 Hydrogen absorption curves of TiVNbTa refractory high-entropy alloy(a)hydrogen absorption kinetics curves;(b)hydrogen absorption pressure curves
通常,温度和压力的增加有利于氢原子的扩散,提高金属吸氢速率,但是当温度较高时,反而会促进金属氢化物的分解。故温度较高会阻碍金属吸氢的进一步进行,从而降低吸氢量。对于TiVNbTa 难熔高熵合金粉末而言,当温度升高至423 K 以上时,吸/放氢平衡向着金属氢化物分解释放的方向进行,所以温度越高,平衡吸氢量越小。
金属的吸氢步骤分别为物理/化学吸附、表面渗透、扩散和形成氢化物[18],每一步都会对吸氢动力学产生影响,成为吸氢动力学的限速步骤,而每个步骤都有其相应的动力学模型。为了进一步研究TiVNbTa 难熔高熵合金的吸氢机理,采用动力学机制方程拟合法对TiVNbTa 难熔高熵合金的吸氢动力学机制进行分析。对TiVNbTa 难熔高熵合金粉末在423,573 K 和723 K 温度的恒温吸氢动力学曲线使用Johnson-Mehl-Avrami(JMA),Power law,Zero-order,First-order,Second-order,Third-order 等主要动力学机制方程进行拟合。经拟合后只有JMA 方程的R2均大于0.99,表明JMA 方程能较好地描述合金粉末的吸氢反应,合金的吸氢反应主要受控于氢化物的形核及长大过程[19]。表1 为Johnson-Mehl-Avrami(JMA)方程 ([-ln(1-α)]1/n=kt)动力学拟合结果,其中α为反应分数;n为反应级数;k为速率常数;t为反应时间,拟合范围为0.15<α<0.95。根据JMA 方程拟合直线,可以得出在不同温度下合金吸氢过程的反应级数n和速率常数k,拟合结果如图5 所示。从图5 中可以看到,温度在423,573 K 和723 K 时,合金的氢化反应只有一个阶段。张诚[20]通过动力学机制方程拟合法对TiZrNbTa 合金的吸氢动力学机制进行分析,分析结果表明,TiZrNbTa 合金的吸氢动力学曲线均遵循JMA 方程。由此可知,TiVNbTa 和TiZrNbTa 高熵合金的吸氢动力学机制相似。

表1 TiVNbTa 难熔高熵合金吸氢动力学机制拟合结果(JMA 方程)Table 1 Fitting results of hydrogen absorption kinetics mechanism of TiVNbTa refractory highentropy alloy(JMA model)

图5 吸氢动力学机制方程拟合结果Fig.5 Fitting results of hydrogen absorption kinetic mechanism equation
由经典相变动力学理论可知,反应级数n主要根据吸放氢动力学曲线开始阶段所对应的拟合直线来计算[21]。当温度从423 K 增加至723 K 时,n值分别为0.44,0.53 和0.57,与0.5 相近,说明氢化反应模型为瞬时形核的扩散控制生长,随着温度的升高,氢化物的起始形核速率增加。在不同温度下,TiVNbTa 难熔高熵合金氢化反应第一阶段的k值随着温度的升高而降低,表明吸氢速率随着温度的升高而降低。根据吸氢速率常数k可以获得TiVNbTa 难熔高熵合金的吸氢表观活化能,由阿伦尼乌斯(Arrhenius)关系,即k=k0×exp(-Ea/RT),其中,k0为系数;Ea为表观活化能,J/mol;R为气体常数,8.31 J/(K∙mol);T为绝对温度,K。通过lnk对1000/T作图并拟合直线,如图6 所示,由拟合直线的斜率可求得TiVNbTa 难熔高熵合金的吸氢表观活化能Ea=-21.87 J/mol。
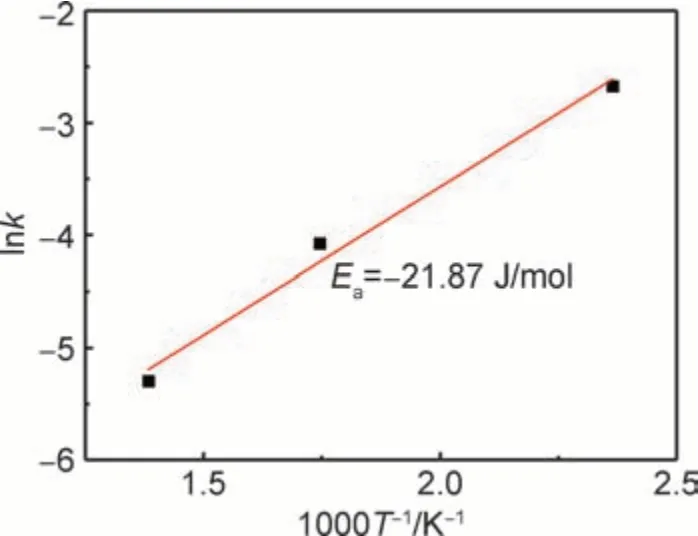
图6 吸氢反应中lnk 与1000T-1的关系曲线Fig.6 Relationship curve between lnk and 1000T-1 in hydrogen absorption reaction
2.3 放氢动力学
图7 (a)为氢化后TiVNbTa 难熔高熵合金粉末在423,573 K 和723 K 的恒温放氢动力学曲线,可以看到,曲线初始阶段斜率随着温度的增加而增加,表明随着温度升高,合金的放氢速率升高,但获得平衡放氢量的时间延长。当温度为423,573 K 和723 K 时,分别需要约80,150 s 和500 s 后才可达平衡放氢量,平衡放氢量(质量分数)分别为0.08%,0.15%和0.32%。可以看到,放氢反应时间低于吸氢反应时间,这是由于在吸氢过程中不断形成氢化物阻碍氢原子在合金中进一步扩散,而在放氢过程中,位于合金表层的氢化物分解时产生的氢原子可直接穿过合金表层释放,氢化物对氢原子扩散的阻碍作用小,氢原子在合金表层的扩散速率远大于在氢化物相中的扩散速率。同时,合金在3 个温度下放氢量远低于吸氢量,均没有彻底放氢。这是因为初始放氢的氢压较低,随着放氢过程的持续进行,在放氢温度的影响下,氢化物分解成氢原子,导致反应室内氢压持续增加,如图7(b) 所示,同时氢原子又不断吸附在合金表面,与合金形成固溶体,直至合金的放氢过程与环境条件达到动态平衡。因此,合金不会彻底放氢,放氢总量受到抑制,想要彻底放氢则需要继续降低环境氢压。
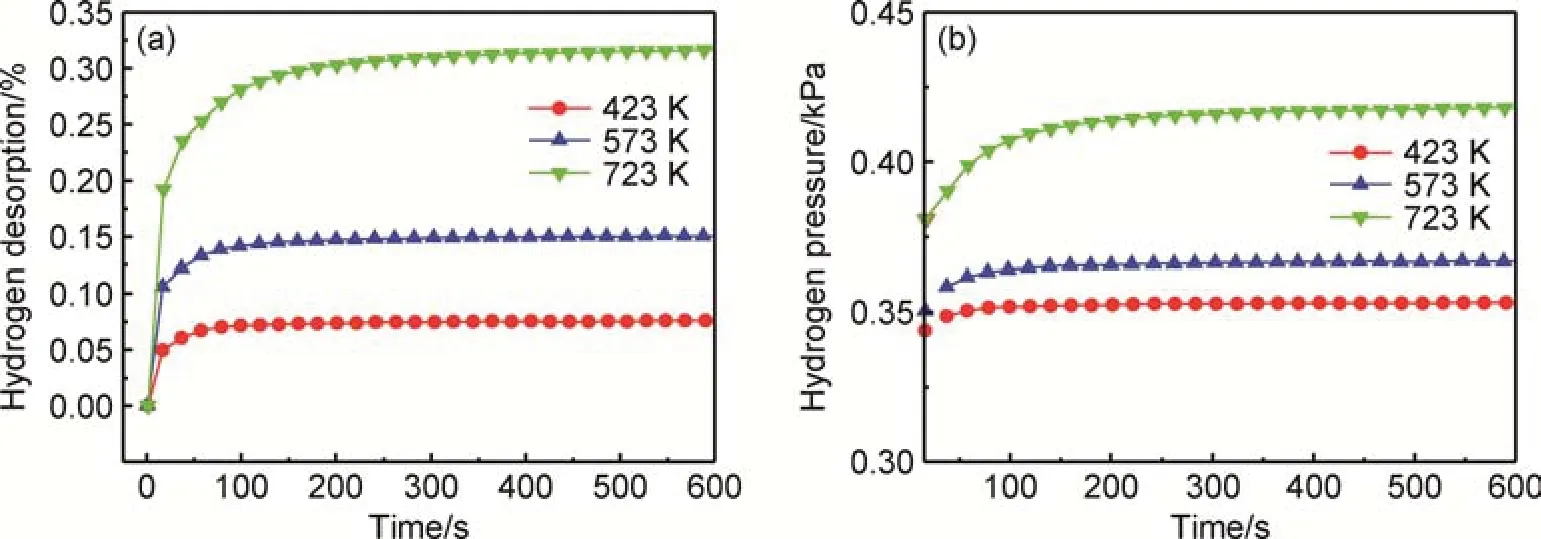
图7 氢化TiVNbTa 难熔高熵合金在不同温度下的放氢曲线(a)放氢动力学曲线;(b)放氢压力曲线Fig.7 Dehydrogenation curves of hydrogenated TiVNbTa refractory high-entropy alloy at different temperatures(a)dehydrogenation kinetics curves;(b)dehydrogenation pressure curves
与吸氢动力学机制分析一致,采用不同动力学机制方程拟合法对氢化后TiVNbTa 难熔高熵合金粉末在不同温度的恒温放氢动力学曲线进行线性拟合,拟合范围为0.15<α<0.95。通过线性拟合结果分析,二级速率方程([1/(1-α)]-1=kt)对应拟合直线的R2均大于0.96,表明该方程为最适合的放氢动力学模型,即放氢动力学机制主要受控于氢化物的浓度含量,拟合结果如图8 所示,拟合参数见表2。当温度从423 K 增加至573 K 时,速率常数k值明显增大,表明TiVNbTa 难熔高熵合金的放氢速率对温度较为敏感,氢化物分解速率随着温度升高而提高,从而提高放氢速率,合金释放氢气也更加容易。而温度从573 K 继续增加至723 K 时,速率常数k值变化不大,表明两者在合金放氢过程的起始阶段放氢速率相近。根据Arrhenius 关系对lnk和1000T-1作图并拟合直线,拟合结果如图9 所示。由拟合直线斜率求得TiVNbTa 难熔高熵合金的放氢表观活化能Ea=8.67 J/mol。

表2 TiVNbTa 难熔高熵合金放氢动力学机制拟合结果(二级速率方程)Table 2 Fitting results of dehydrogenation kinetics mechanism of TiVNbTa refractory high-entropy alloy (Second-order model)
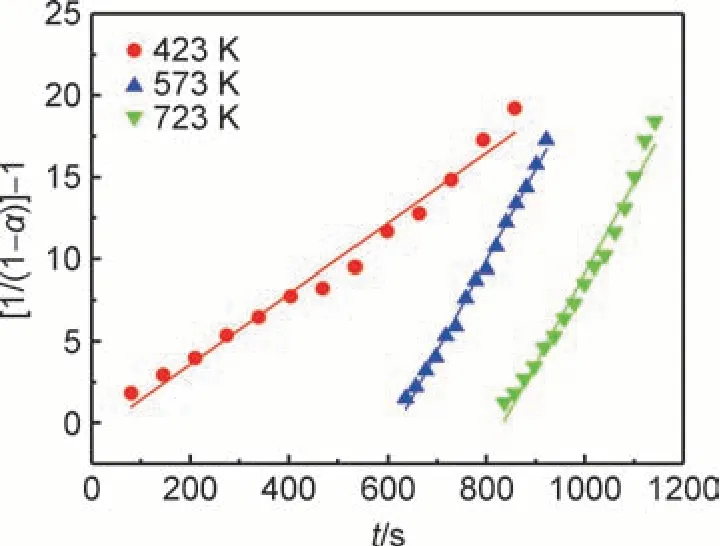
图8 放氢动力学机制方程拟合结果Fig.8 Fitting results of dehydrogenation kinetic mechanism equation
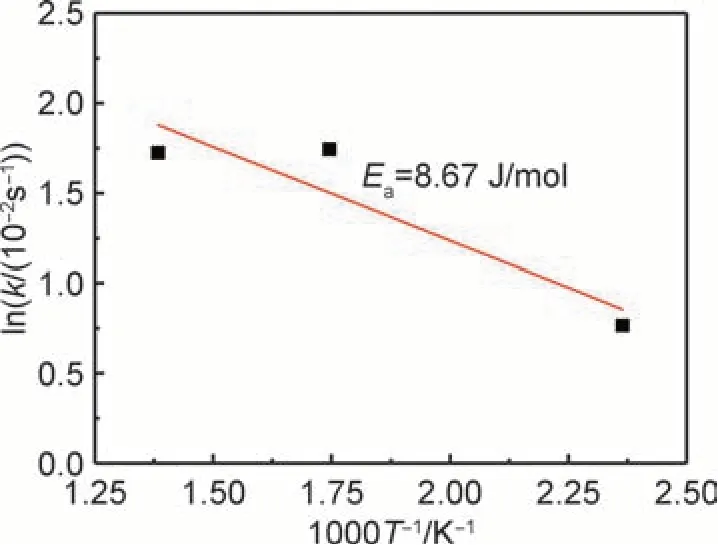
图9 放氢反应中lnk 与1000T-1的关系曲线Fig.9 Relationship curve between lnk and 1000T-1 in dehydrogenation reaction
3 结论
(1)TiVNbTa 难熔高熵合金吸氢前为单相BCC结构,吸氢后生成TiH1.971,Nb0.696V0.304H 和Nb0.498V0.502H23 种氢化物新相,完全放氢后的合金再次恢复单相BCC 结构。DSC 曲线表明,氢化后的TiVNbTa 难熔高熵合金在放氢过程中,合金氢化物的分解需要三步反应,分别在519,593 K 和 640 K 发生氢化物分解。
(2)活化后的TiVNbTa 难熔高熵合金在1.8 MPa的初始压力,423,573 K 和723 K 温度下均可以迅速吸氢,无孕育期出现,并且随着温度的升高,吸氢速率和平衡吸氢量降低。合金在3 个温度下经过约280,500 s和900 s 后达到平衡吸氢量,平衡吸氢量(质量分数)分别为0.96%,0.90% 和0.77%。合金的吸氢动力学曲线遵循JMA 方程,吸氢动力学机制主要为瞬时形核的扩散控制生长,吸氢的表观活化能Ea为-21.87 J/mol。
(3)氢化后的TiVNbTa难熔高熵合金在423,573 K和723 K 温度下,经过约80,150 s 和500 s 后达到平衡放氢量,平衡放氢量(质量分数)分别为0.08%,0.15%和0.32%。随着温度的升高,平衡放氢量增加,放氢速率提高。放氢动力学曲线遵循二级速率方程,放氢动力学机制主要受控于氢化物的浓度含量,放氢的表观活化能Ea为8.67 J/mol。
