Milling degree affects the fermentation properties of rice: perspectives from the composition of nutrients and gut microbiota via in vitro fermentation
2024-01-24YuZhngFnLiShutongPnBingBiKiHungSenLiHongweiCoTinXieJinXieXioGun
Yu Zhng, Fn Li, Shutong Pn, Bing Bi, Ki Hung, Sen Li,Hongwei Co, Tin Xie, Jin Xie, Xio Gun,
a School of Health Science and Engineering, University of Shanghai for Science and Technology, Shanghai 200093, China
b National Grain Industry (Urban Grain and Oil Security) Technology Innovation Center, Shanghai 200093, China
c Nutrition & Health Research Institute Co. Ltd., COFCO Corporation, Beijing 102209, China
d China Grain Wuhan Scientif ic Research & Design Institute Co. Ltd., Wuhan 430079, China
Keywords: Rice processing Milling Whole grains Gut microbiota
ABSTRACT Fermentation substrates of rice with different milling degrees (MDs) were prepared and fermented with human feces to compare their fermentation properties and effects on gut microbiota. MD 0s, MD 5s and MD 60s represented brown rice, moderately-milled rice and white rice, respectively. After in vitro fermentation,the MD 5s group showed higher starch utilization, compared with the MD 0s and 60s groups evaluated by Fourier transform infrared spectrometer, and confocal laser scanning microscope. Effects of fermentation substrates of rice with different MDs on gut microbiota were evaluated by 16S rDNA sequencing. All the sample groups reduced the pH and produced short-chain fatty acids (SCFAs) and branched-chain fatty acids. The MD 5s group exhibited higher α-diversity than the MD 0s and 60s groups. Abundances of Phascolarctobacterium, Blautia and norank_f_Ruminococcaceae were higher in the MD 0s and 5s groups, compared with the MD 60s group. These bacteria were also positively correlated with the SCFAs production via Spearman correlation analysis. In vitro culture assay revealed that fermentation substrates of MD 0s and 5s promoted the growth of two probiotics (Akkermansia muciniphila and Bif idobacterium adolescentis). Our results showed that moderate milling might be an appropriate way to produce rice products with richer nutrients and better fermentation properties.
1. Introduction
Rice is the staple food for more than half of the world’s population. Polished rice, also known as white rice, is the most popular rice product due to its better palatability. However, rapid starch digestion and the loss of nutrients during milling of white rice have brought some health concerns, such as type 2 diabetes and insulin resistance if being consumed for a long time[1]. Thus, wholegrains, such as brown rice has caught public attention and were reported to exhibit various health benef its[2]. However, relatively high price, poor mouthfeel and cooking property limit the prevalence of brown rice. Recent studies have shown that appropriate processing might be effective in maintaining the nutrients and improving the rheological property of whole-grains[3]. Our previous study has shown that moderately-milling maintained most of the nutrients, slowed the starch digestion, and improved the cooking property of brown rice[4].However, the fermentation properties of moderately-milled rice and its interaction with gut microbiota was scarcely reported and awaits more investigation.
After digestion, most part of the white rice (mainly composed of starch) is absorbed. As for brown rice or other whole grains, dietary fiber and some nutrients binding to dietary fiber, such as proteins and polyphenols might not be easily digested and absorbed and would further reach the colon and interact with gut microbiota[5-6]. After interacting with gut microbiota, some metabolites, such as shortchain fatty acids (SCFAs) and branched-chain fatty acids (BCFAs)would be produced and further affect gut health and host metabolism acting as signaling regulators[7]. Milling can remove the bran layers,including pericarp, testa and aleurone layer of cereal grains. Different degrees of milling leave different proportion of the bran layer and affect the binding among the nutrients. This would further affect their digestion properties[4]. Thus, the composition of rice with different milling degrees (MDs) would be different after digestion and absorption, and leave with different fermentation substrates for gut microbiota for fermentation and utilization[8]. This would further affect gut microenvironment and gut health. Although whole grains have been widely studied in terms of their effects on gut health,how gut microbiota responds to rice with different MDs is yet to be investigated.
Thus, fermentation substrates of rice with different MDs were prepared in the present study and their compositions were determined chemically. Feces from healthy volunteers were collected and fermented with fermentation substrates of rice with different MDs.Changes of compositions were observed by Fourier transform infrared spectrometer (FT-IR) and confocal laser scanning microscope(CLSM) before and afterinvitrofermentation. The effects of fermentation substrates of rice with different MDs on the composition of gut microbiota were determined by 16S rDNA sequencing.Metabolites, such as SCFAs and BCFAs afterinvitrofermentation were determined and quantified by liquid chromatograph-mass spectrometer (LC-MS). Spearman correlation analysis was applied to look for the key bacteria contributing to the production of these metabolites. Afterwards, different types of common bacteria were further cultured with fermentation substrates of rice with different MDs to verify the results of 16S rDNA sequencing. Our results would fill the gap of the fermentation properties of moderately-processed cereal grains and provide guidance for the rice processing industry.
2. Materials and methods
2.1 Materials
Wuchang rice grains (Japonica rice line) were purchased from Caiqiao Rice Industry Co., Ltd. (5448130-2KG, Wuchang Country,Heilongjiang province, China). All the chemicals used were at analytical grade.
2.2 Preparation of the fermentation substrates
Rice was cooked and was prepared as cooked rice flour according to our previous study[4]. Fifty-four grams of cooked rice flour was dissolved in 378 mL of phosphate buffer (20 mmol/L, pH 6.9,containing 10 mmol/L of NaCl) at 37 °C. 9 mL ofα-amylase solution(28.7 U/mL, containing 1 mmo/L of CaCl2) was added into the solution to simulate oral digestion for 15 min. The pH was adjusted to 2.0, and 9 mL of pepsin (300 U/mL, containing 15.5 mmol/L of NaCl) was added to simulate gastric digestion for 30 min at 37 °C.Then, the pH was adjusted to 6.9, and 9 mL of pancreatin (4.5 mg in phosphate buffer) and 9 mL of amyloglucosidase solution were added to simulate intestinal digestion at 37 °C for 6 h. The digestion solution was heated in the boiling water for 2 min to stop the digestion. In order to exclude the effects of the absorbed components on the following fermentation, the digestion solution was dialyzed for 26 h (6–8 kDa). Then, the remaining solution in the dialysis bag(unabsorbed components) was freeze-dried and collected for further use as the fermentation substrates.
2.3 Determination of starch, protein and fiber in the fermentation substrates
The starch content was determined by the total starch (AA/AMG) assay kit according to the manufacturer’s instruction. The protein content was determined according to AACC Method 46-12.01. The fiber content was determined by the Total Fiber Assay kit(K-TDFR, Megazyme International, Wicklow, Ireland) according to the manufacturer’s instruction.
2.4 In vitro fermentation of the fermentation substrates of rice with different MDs
The process ofinvitrofermentation was carried out based on our previous study[9]. The fermentation substrates were sterilized by heating in a 90 °C water bath for 30 min and then cooled to room temperature. Two hundred milligram of MD 0s, MD 5s, MD 60s samples,fructo-oligosaccharide (FOS) were added to the 50 mL vials. The culture broth without adding carbon source was used as the blank group.
Four healthy volunteers (3 males, 1 female) were selected according to previous studies[9-11]. Their BMI values were between 18.5 and 25 kg/m2. They have not been taking antibiotics for at least 3 months prior to participating in this study. They have no intestinal disease and are all informed and agree with the research[9]. The feces of the volunteers were collected within 1 h, and 5 g of each was quickly weighed and mixed evenly. The mixed feces were added to carbonic acidphosphate buffer at a ratio of 1:4 (m/V). The filtrate was collected and quickly placed in an anaerobic environment for the next step.
Sixteen milliliter of autoclaved carbonic acid-phosphate buffer and 4 mL of fecal filtrate were added and mixed with the fermentation substrates. The mixtures were cultured anaerobically under N2/H2/CO2(90:5:5,V/V) at 37 °C. Tested samples were taken at 0, 6, 12, and 24 h,respectively for the following investigation.
2.5 Observation of samples by FT-IR and CLSM
Infrared spectra of samples before and afterinvitrofermentation were analyzed with an FT-IR spectroscopy instrument (NicoletiS50,Thermo Scientific, UK). The dried powder was weighed and mixed with an appropriate amount KBr (1:100,m/m). Then, it was ground into homogeneous powder and pressed into transparent discs. Spectral wavenumbers were collected in the range of 400–4 000 cm–1. The mixture was scanned for 32 times with a resolution of 4 cm–1.
The starch utilization of the samples before and afterinvitrofermentation was observed by CLSM based on a previous study with slight modifications[12]. 10 mg of the sample was uniformly dispersed in a mixture of 15 μL of 1 mol/L CH3BNNa and 15 μL of freshly prepared 8-amino-1,3,6-pyrenetrisulfonic acid (APTS). The mixture was incubated and stained at 30 °C for 18 h. Then, the sample was washed 5 times with 1 mL of distilled water, and finally resuspend in 20 μL of 50% glycerol and observed under a CLSM (Zeiss LSM900,German). The fluorescent intensity and staining area were analyzed by ImageJ for quantification.
2.6 Analysis of gut microbiota by 16S rDNA sequencing
Afterinvitrofermentation, fecal samples were collected and used for bacterial 16S rDNA sequencing as reported in our previous study[13]. Microbial community genomic DNA was extracted using TruSeqTMDNA Sample Prep Kit (Omega Bio-tek, Norcross, GA,USA) according to manufacturer’s instruction. The 16S rRNA comprising the V3–V4 regions was amplified. The sequencing was conducted by Marjorbio Bio-Pharm Technology Co. Ltd. using Illumina MiSeq PE300 platform/NovaSeq PE250 platform (Illumina,San Diego, USA). The raw 16S rRNA gene sequencing reads were demultiplexed, quality-filtered by fastp version 0.20.0 and merged by FLASH version 1.2.7. Operational taxonomic units (OTUs) with 97% similarity cutoff were clustered using UPARSE version 7.1, and chimeric sequences were identified and removed. The taxonomy of each OTU representative sequence was analyzed by RDP Classifier version 2.2 against the 16S rRNA database (eg. Silva v138) using the confidence threshold of 0.7. Alpha diversity indices, relative abundance of phyla, principal component analysis (PCA) and linear discriminant analysis (LDA) Effect size (LEfSe) analysis were assessed.
2.7 Analysis of SCFAs
Extraction and quantification of SCFAs were done as previously reported with slight modification[14]. 50 μL of fermentation supernatant was mixed with 100 μL of acetonitrile (ACN), and was sonicated at 5 °C for 30 min. After centrifugation at 13 000 r/min at 4 °C for 15 min, 40 μL of the supernatant was mixed with 20 μL of 200 mmol/L of 3-nitrophenylhydrazine hydrochloride in 50% ACN and 20 μL of 120 mmol/L of EDC-6% pyridine in 50% ACN. The mixture was reacted at 40 °C for 30 min, and then diluted with 50%ACN for further analysis.
A LC-MS/MS ExionLC AD system was used for analysis of SCFAs. 10 μL of sample was injected onto a Waters BEH C18(150 mm × 2.1 mm, 1.7 μm) column at 40 °C. Mobile phase A was 0.1% formic acid and mobile phase B was 0.1% formic acid-ACN.The flow rate was 0.6 mL/min. The gradient elution profile was as follows: 10%–20% B (0–4 min), 100% B (4–4.1 min), 100% B(4.1–7.5 min), 10% B (7.5–7.6 min). SCFAs concentration of samples was calculated based on the retention time and the integrated peaks with the standard curves of acetic acid, propionic acid, butyric acid,isobutyric acid, valeric acid and isovaleric acid.
2.8 Culture of 4 types of common bacteria with the fermentation substrates of rice with different MDs
Culture of 4 types of common bacteria was done based on a previous study with slight modification[15]. The second-generation bacteria were inoculated at 1% (V/V) into 10 mL of the medium supplemented with the corresponding fermentation substrates (MD 0s, 5s and 60s). Bacteria were grown under anaerobic conditions at 37 °C, and the control gas ratio was N2/H2/CO2(90:5:5,V/V).The test tube was taken out at the corresponding detection time point, and was vortexed for 10 s and then left to stand for 5 min. The growth of bacteria was assessed by the value of OD600nm.Escherichia coli,BifidobacteriumadolescentisandBacteroides acidifacienswere assessed within 48 h.Akkermansiamuciniphilawas assessed within 168 h.
2.9 Statistical analysis
The data from this study are presented as the mean ± standard deviation (SD) for at least three replicates of each sample.Regression analyses and other statistical analyses were performed using Microsoft Excel 2007 and GraphPad Prism (version 7.0). The means were compared using ordinary one-way analysis of variance(ANOVA). Statistically significant differences were set atP< 0.05,0.01 and 0.001.
3. Results and discussion
3.1 Composition of rice fermentation substrates with different MDs before in vitro fermentation
The rice sample used in the present study was Wuchang rice grains (Japonica rice line, Wuyoudao 4). We investigated three typical types of rice in China in our previous study[4], i.e. red rice grains (Indica rice line, Ganwanxian 33), Wuchang rice grains(Japonica rice line, Wuyoudao 4) and Ningxia rice grains (Japonica rice line, Fuyuan 4). We found that the starch digestion and textural properties of Wuchang rice at different milling degrees (MD 0s, 5s and 60s) showed significant differences, while the other two types of rice did not. So, we continued studying the fermentation properties of Wuchang rice in the present study. Also, Wuchang rice grows in the north-east region with excellent natural conditions in China. Thus,Wuchang rice has become one of the representatives of high-quality rice and shows great popularity in China[16].
Before reaching the colon, food matrix has been digested in stomach and a part of which has been absorbed in small intestine.To reflect the behaviors of food matrix in gastrointestinal tract, we prepared the rice fermentation substrates, which were the lyophilized samples afterinvitrodigestion and dialysis of cooked rice with different MDs. Table 1 shows that composition of rice fermentation substrates with different MDs. Results showed that total starch contents were around 46% in MD 5s and MD 60s groups, while was around 40% in MD 0s. On the contrary, the content of total fiber decreased significantly from MD 0s (20.67%) to MD 60s (12.51%)(P< 0.05). Total starch and total fiber accounted for the most composition of rice fermentation substrates. Their differences in content and composition would cause differences in the composition of gut microbiota that utilized them for living.

Table 1 Total starch, protein and dietary fiber contents of rice fermentation substrates with different MDs.
3.2 Microstructure and changes in starch utilization of fermentation substrates of rice with different MDs after in vitro fermentation
The CLSM images and FT-IR spectra of rice with different MDs before and afterinvitrofermentation were shown in Fig. 1. Due to the existence of incompletely digested starch and fiber in the fermentation substrates (Table 1), changes of the absorption peaks at 1 047, 1 022,995 cm–1were observed, and these were associated with changes of starch structures[17]. After fermentation, the number and shape of absorption peaks changed obviously from 800–1 300 cm–1. Bands around 1 150 and 1 030 cm–1were assigned to C–O–C stretching vibration in cellulose and semi-cellulose[18], and their disappearance indicated that gut microbiota utilized these nutrients duringinvitrofermentation. The ratio of peak intensities of 1 047/1 022 cm–1to 995/1 022 cm–1is considered as the reflection of degree of order (DO)and degree of the double helix (DD) of starch-based samples[19]. Afterinvitrofermentation, DO and DD values of all groups increased to some extent (Table 2), and MD 5s showed the highest levels among all (P< 0.05). This might be due to the hydrolysis of the amorphous structure of starch by enzymes secreted by gut microbiota, and thus increased the DO of samples[20]. Also, the peak intensities of bands around 3 000–3 600 cm–1representing –OH stretching vibration and bands around 1 640 cm–1representing water attached to the amorphous part of starch increased significantly, indicating the molecular rearrangement of starch duringinvitrofermentation[21].
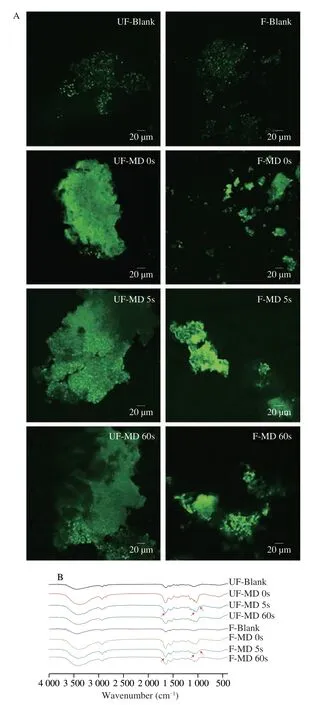
Fig. 1 CLSM images (A) and FT-IR spectra (B) of rice with different MDs before and after in vitro fermentation. UF, unfermented sample, F, fermented sample (Scale bar: 20 μm).

Table 2 Changes in starch utilization, DO, DD and fluorescent intensity of rice with different MDs before and after in vitro fermentation.
APTS as a specific fluorescent dye for starch can reflect the internal structure and the distribution of amylose and amylopectin and starch granules. By binding to the reducing end of starch molecules,more APTS can be easily bind to amylose compared with amylopectin because amylose has much more reducing ends compared with amylopectin, and thus has stronger fluorescent intensity[22]. It has been proposed that bright and uniform fluorescence might be attributed to the compact structure of starch granules[23]. Fig. 1A shows that compared with UF-MD 60s, UF-MD 0s and UF-MD 5s had stronger fluorescent intensities, indicating that milling might cause unordered structure of starch in rice[21]. Afterinvitrofermentation,fermented samples had stronger fluorescent intensities compared with unfermented samples, which might be due to the existence of highamylose starch and/or amylopectin at low molecular weight[24]. Duringinvitrofermentation, enzymes secreted by gut microbiota hydrolyzed starch and produced more amylose or amylose/amylopectin with shorter chain lengths, which made the structural arrangement more ordered and thus increased the fluorescent intensity. Among the fermented groups, the fluorescent intensity of F-MD 5s was significantly stronger than that of F-MD 0s and F-MD 60s, indicating that the utilization of starch by gut microbiota was higher in MD 5s.This was also in line with the starch utilization shown in Table 2.This might be due to some mechanical damage on rice kernels by moderate milling, which might make gut microbiota easier to utilize the nutrients, compared with MD 0s. Also, compared with MD 60s, a higher content of dietary fiber was reported to enrich the diversity of gut microbiota, and might also promote the process of fermentation[8].Difference in the utilization of starch and other carbon sources might further induce differences in the composition of gut microbiota and related metabolites[20-21].
3.2 Effects of rice with different MDs on the composition of gut microbiota after In vitro fermentation
Milling caused different composition and different structure of components (i.e. starch) of rice samples, which might further affect the utilization by gut microbiota, and thus change their composition reciprocally. The composition of gut microbiota afterinvitrofermentation was determined by 16S rDNA sequencing. The Venn diagram shows that 299 OTUs were shared by all the tested groups(Fig. S1A). The MD 0s and MD 60s groups did not show their unique OTUs, while the MD 5s and FOS (positive control) groups showed 8 and 2 unique OTUs, respectively. Inα-diversity (Fig. S1B), Ace and Chao 1 indexes did not show obvious different among different sample groups. On the other hand, Shannon index reflecting the richness and evenness was significantly decreased and Simpson index reflecting the uniformity of the community was significantly increased afterinvitrofermentation with MD 0s, 5s and 60s groups (P< 0.05).This indicated that rice with different MDs increased the evenness of the gut microbiota[25]. Compared with the MD 0s and MD 60s groups, the MD 5s group had the closest values of the indexes ofα-diversity with those of the FOS group, and the MD 5s group exhibited higherα-diversity taking the Venn diagram and Simpson index into account. Previous studies have shown that the higher the diversity and evenness of the gut microbiota, the lower the sensitivity to gut diseases[26-27]. Based on the above results, the positive effects of MD 5s in maintaining the diversity and evenness of gut microbiota,compared with MD 0s and MD 60s, might be attributed to the differences in the components of fermentation substrates and the utilization of starch.
As shown in Fig. 2A, Bacteroidota, Firmicutes, Proteobacteria,Actinobacteriota, and Desulfobacterota contributed to the most composition of gut microbiota (Fig. 2A). PCA analysis revealed that the initial, blank, FOS and MD 0s, 5s, 60s groups showed obvious separation with each other (Fig. 2B), indicating their differences in the composition of gut microbiota. The MD 0s and MD 5s groups collapsed with each other in the Bray-Curtis distance matrix, while both separated with the MD 60s group obviously (Fig. 2C), indicating that moderately-milled rice (MD 5s) shared similar composition of gut microbiota with brown rice (MD 0s) afterinvitrofermentation.Bacteroidotawas reported to be the major phylum that secreted lyases and hydrolases degrading the main and side chains of polysaccharides and produced degraded products being utilized by other gut microbiota[28-30]. The abundance of Bacteroidota did not show significant differences among the MD 0s, 5s, 60s and FOS groups.This might be due to the similar contents of accessible carbohydrates that can be directly utilized by Bacteroidota in these groups. Besides,other than Bacteroidota, the increased proportion of Firmicutes was reported to be associated with protein fermentation[31]. The proportion of Firmicutes in sample groups was in line with their contents of protein (Table 1). Actinobacteriota containingBifidobacteriumand other probiotics was found to be the richest in the FOS group (1.94%),following by the MD 5s group (1.72%). An increased proportion of Actinobacteriota was also observed in mice after consuming wholegrain products[8,32-33]. Compared with other distinguished phylum,Proteobacteria contains many kinds of pathogens or opportunistic pathogens, which might induce inflammation and chronic colitis[34-35].In treatment groups, the proportion of Proteobacteria was the lowest in the FOS group (2.05%), while, the highest in the MD 0s group (5.12%).
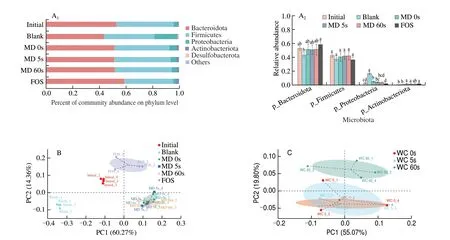
Fig. 2 Effects of rice with different MDs on the composition of gut microbiota after in vitro fermentation. (A1, A2) Changes of microbiota at phylum level.(B) PCA of microbiota among the initial, blank, MD 0s, MD 5s, MD 60s and FOS groups. (C) PCA of microbiota among the MD 0s, MD 5s and MD 60s groups.Results are expressed as mean ± SD (n = 4). Different letters refer to significant difference at P < 0.05. Multiple comparisons were performed by one-way ANOVA followed by Tukey’s test.
3.3 Gut microbiota responded differently to the fermentation substrates of rice with different MDs at genus level
Although at phylum level, the composition of gut microbiota of sample groups did not show obvious difference, their distinguished bacteria at genus level were different (Fig. 3A and S2).Prevotella,Bacteroides,Faecalibacterium,Roseburia,Phascolarctobacterium,Agathobacterand Lachnospiraceae_NK4A136_group contributed to more than 50% of the gut microbiota in MD 0s, 5s, 60s and FOS groups (Fig. 3B). LEfSe analysis was applied to show the distinguished bacteria at genus level of each sample group (Fig. 3C).For MD 0s,Phascolarctobacterium,Escherichia-Shigella, OTU331 and OTU327 were the most distinguished genus. For MD 5s,Agathobacter, OTU439 andEubacterium_rectale_ATCC 33656 were the most distinguished genus. For MD 60s,Prevotella,Roseburia,Lachnospiraceae_NK4A136_group and Lachnospiraceae_bacterium_GAM79 species were the most distinguished genus. In comparison,the distinguished genus in the FOS group wereRuminococcus_torques_group,Blautiaand OTU247.

Fig. 3 Distinguished bacteria at genus level of rice with different MDs after in vitro fermentation. (A) The abundance of 8 types of distinct bacteria at genus level. (B) the composition of microbiota at genus level. (C) LEfSe analysis of the microbiota at genus level, (A1–A8) Phascolarctobacterium, Blautia, Alistipes,Escherichia-Shigella, norank_f_Ruminococcaceae, norank_f_Muribaculaceae, Bacteroides, Comamonas, respectively. Results are expressed as mean ± SD (n = 4).Different letters refer to significant difference at P < 0.05. Multiple comparisons were performed by one-way ANOVA followed by Tukey’s test.
Phascolarctobacteriumas a distinguished bacterium in MD 0s,is widely found in human gastrointestinal tract. Previous studies have shown that it could utilize succinate, produce SCFAs and was associated with the metabolic state of the host[36-37].AgathobacterandEubacterium_rectale_ATCC 33656 were distinguished bacteria in MD 5s. The number of studies aboutAgathobacteris quite scarce and requires much more investigation.Eubacterium_rectale_ATCC 33656 belonging to Firmicutes, is one of the most abundant butyrateproducing bacteria in human gut[38]. It consumed a limited range of substrates, including starch, and thus diets rich in resistant starch could promote the abundance ofE. rectale. Decreased abundance ofE. rectalewas observed in some diseases, such as obesity,inflammatory bowel disease and diabetes, etc.[39]. The fermentation substrate of MD 5s contained more starch compared with that of MD 0s and more dietary fiber compared with that of MD 60s.This might promote the growth ofEubacterium rectale, which contributed to higher starch utilization in the MD 5s group (Table 2).Eubacterium rectalewas also found to be increased in the gut of mice after consuming whole grains[33].PrevotellaandRoseburiawere distinguished bacteria in MD 60s, and both of them have been paid much attention in terms of their effects on host health.Prevotellacontributing to a large proportion ofBacteroidota, was found to be commonly associated with non-western diets and diets rich in carbohydrates, resistant starch and fiber. It has the potential to degrade complex polysaccharides and produce SCFAs from arabinoxylans and FOSinvitro[40].Roseburiabelonging to Firmicutes, is also a butyrate-producing bacteria and has shown the promise being used as a novel probiotics[41]. Their rich abundance might further explain the production of SCFAs and the high starch utilization rate in the MD 60s group.
3.4 Production of SCFAs and BCFAs and their correlation with gut microbiota after in vitro fermentation of rice with different MDs
Duringinvitrofermentation, as the accumulation of metabolites,such as SCFAs increased, the pH of the culture medium would change and further affect the growth and the composition of gut microbiota[42].Thus, the pH value of the culture medium can be considered as an indicator of the utilization of samples and the degree of fermentation.Fig. 4A shows that the pH of all groups decreased dramatically within the first 6 h, which might be due to the production of organic acids[9].From 6 till 24 h, the pH values remained constant. After a 24-hinvitrofermentation, the reduction of pH values in FOS, MD 0s, MD 5s and MD 60s groups were 1.99, 0.95, 0.98 and 0.99, respectively.Appropriate reduction of pH might promote the growth of some probiotics, and inhibit the growth of some harmful bacteria[43]. Since the reduction of pH was similar among the sample groups, detailed contents of total and individual SCFAs were determined by LC-MS.
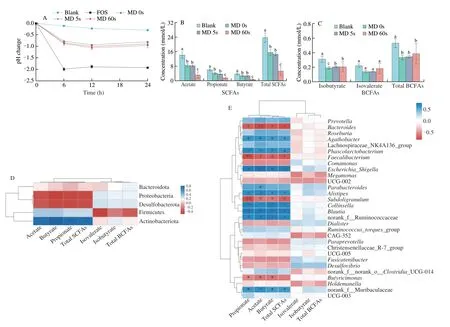
Fig. 4 Production of SCFAs and BCFAs and their correlation with gut microbiota after in vitro fermentation of rice with different MDs. (A) The pH, contents of (B) SCFAs and (C) BCFAs. Results are expressed as mean ± SD (n = 4). Different letters refer to significant difference at P < 0.05. Multiple comparisons were performed by one-way ANOVA followed by Tukey’s test. Spearman correlation analysis among SCFAs, BCFAs and microbiota at (D) phylum and (E) genus levels. Significant correlation at +P < 0.1, *P < 0.05, and **P < 0.01.
Fig. 4B and C show the contents of SCFAs and BCFAs in different groups. Carbohydrates, proteins and amino acids can be fermented by gut microbiota to produce SCFAs and BCFAs. Fig. 4B shows that the MD 0s group had the highest production of SCFAs(23.81 mmol/L), followed by the MD 5s (15.56 mmol/L) and MD 60s(14.57 mmol/L) groups. This might be due to the existence of dietary fiber in the fermentation substrates in each group (Table 1), and the production of SCFAs was in consistency with the content of dietary fiber. Resistant starch as a type of dietary fiber, its fermentation was reported to promote the production of butyrate[44]. Our previous study has investigated the content and type of resistant starch of rice with different MDs duringinvitrodigestion, and results showed that MD 0s and MD 5s had the highest contents of resistant starch[4]. The resistant starch in MD 0s was mainly I and V types, 5s of milling did not affect the content of the original resistant starch of brown rice. Besides, since MD 0s was not milled, it had a higher content of fiber in cell wall, which might also produce SCFAs duringinvitrofermentation[31]. Acetate, as the major SCFAs produced from rice with different MDs, was reported to show promising effects in various diseases, such as obesity, diabetes, inflammatory bowel diseases and neurological disorders, etc.[7]. Besides, BCFAs was generally produced from the fermentation of branched amino acids. After a 24-h fermentation, the MD 0s group had the highest contents of isovalerate and isobutyrate, while other groups shared similar contents of total BCFAs. Branched-chain ratio (BCR) referred to as the ratio of BCFAs to straight-chain fatty acids was reported to reflect the degree of protein fermentation[45]. The MD 60s group had the highest BCR(2.37%), followed by MD 0s (2.25%) and MD 5s (2.16%). Other than BCFAs, some nitrogen-containing metabolites, which might bring some negative effects on colon might also be produced during protein fermentation[46].
SCFAs could act as the regulators of the host metabolism. For example, acetate could be provided as energy source for the host[31];propionate was reported to reduce the biosynthesis of cholesterol;and butyrate could help to maintain the integrity of gut barrier and improve gut immunity[47]. Spearman correlation analysis was applied to analyze the correlation between the abundance of gut microbiota and the contents of different SCFAs and BCFAs. At phylum level(Fig. 4D), the contents of SCFAs were positively correlated with Actinobacteriota (P< 0.05), while were negatively correlated with Proteobacteria and Desulfobacterota. Previous studies showed Actinobacteriota was involved in the production of SCFAs[48]. The MD 0s,5s and the FOS groups showed higher Actinobacteriota proportion, which might explain their higher contents of SCFAs (Fig. 4B).
At genus level (Fig. 4E), the contents of SCFAs were positively correlated withPhascolarctobacterium,Escherichia-Shigella,Alistipes,Collinsella,Blautiaand no_rank_f_Ruminococcaceae (P< 0.05), while were negatively correlated withBacteroides,Faecalibacterium,Subdoligranulum(P< 0.05).Phascolarctobacteriumwas reported to participate in the production of propionate via succinic acid cycle[49]. Its higher abundance in the MD 0s might explain the higher content of propionate in the MD 0s group as well.Escherichia-Shigellaas an opportunistic pathogen,was found to be increased after fermentation with rice with different MDs, since it might utilize the fermentation substrates of each group as the low-molecular carbohydrate sources to grow[34]. Although fermentation substrates of rice with different MDs increased the abundance ofEscherichia-Shigella, such level (4% in the MD 0s group) was still within the normal level of healthy people[50].Blautiais a type of typical bacteria that can ferment carbohydrates to produce acetate and was found to be enriched in the feces after whole-grain supplementation[51]. A relatively higher abundance ofBlautiain the MD 0s and MD 5s groups might explain relatively higher contents of acetate in these two groups.Bacteroideswas reported to be able to utilize amino acids and might also produce some toxic metabolites, such as indole and skatole[52]. Fermentation with rice with different MDs reduced the abundance ofBacteroides, compared with the blank group, and thus it was negatively correlated with the production of SCFAs.
Thus, gut microbiota utilized the fermentation substrates of rice with different MDs differently and caused differences in metabolites(SCFAs and BCFAs) and the composition of gut microbiota as well.MD 0s and MD 5s promoted the growth of acid-producing bacteria and showed higher levels of SCFAs.
3.5 Effects of fermentation with rice with different MDs on the growth typical bacteria species
To further verify the results of 16S rDNA, four typical species of gut bacteria,A.muciniphila,B. acidifaciens,E. coliandB.adolescentis, were further cultured with the supplementation of the fermentation substrates of rice with different MDs (Fig. 5).A.muciniphilabelonging to Verrucomicrobiota is regarded as a nextgeneration probiotic that can degrade mucins for living and produce SCFAs to promote gut health[53]. After 144 h, compared with the control group, supplementation with the fermentation substrate of MD5s promoted the growth ofA.muciniphilathe most (P< 0.05),followed by the MD 0s and MD 60s groups. This suggested that compared with brown rice (MD 0s) and white rice (MD 60s),moderately-milled rice (MD 5s) could promote the growth ofA.muciniphila. As forB. acidifaciensbelonging toBacteroidota,it grew dramatically after 12 h. However, supplementation with the fermentation substrate of MD 0s, 5s and 60s delayed the growth ofB. acidifaciens, which was in consistency with the results of 16S rDNA sequencing (Fig. 3C). Studies aboutB. acidifacienswere quite controversial. Some reported its effects on metabolizing isoflavone, promoting IgA production and preventing obesity,while, some reported its association with liver disease[54].E. colibelonging toEscherichia-Shigellagenus was significantly higher after supplementation with the fermentation substrate of MD 0s,while significantly lower after supplementation with the fermentation substrate of MD 5s and 60s, compared with the control group(P< 0.05). Theinvitroculture result differed from that of the 16S rDNA sequencing in term of this bacteria (Fig. 3C), indicating the necessity to investigate the interaction between diet intervention and gut microbiota at a deeper level (species or strain level). Besides,B.adolescentisbelonging toBifidobacteriumgenus, Actinobacteriota phylum is a probiotic that has attracted great attention and been investigated about its health benefits[54]. Results showed that supplementation with the fermentation substrate of rice at different MDs all promoted the growth ofB.adolescentisto different extents.At 32 h, the growth ofB.adolescentiswas the highest in the MD 0s group, followed by the MD 5s and MD 60s groups. This was also in line with some previous studies that whole-grain diet promoted the abundance ofB.adolescentisin human[33].
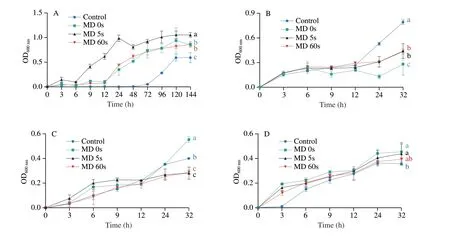
Fig. 5 Growth curves of four types of bacteria species after fermenting with rice with different MDs in vitro. (A) A. muciniphila; (B) B. acidifaciens; (C) E. coli;(D) B. adolescentis. Different letters refer to significant difference at P < 0.05. Multiple comparisons were performed by one-way ANOVA followed by Tukey’s test.
4. Conclusion
The present study investigated the effects of the fermentation substrates of rice with different MDs on the composition of gut microbiota and the production of metabolites (SCFAs and BCFAs),and the changes of the composition of the fermentation substrates before and afterinvitrofermentation. Afterinvitrofermentation, the MD 5s group showed the highest DO value and fluorescent intensity via FT-IR and CLSM, and had a higher starch utilization rate,compared with the MD 0s and MD 60s groups. After the fermentation substrates were degraded and utilized by gut microbiota, the pH values significantly reduced. Combining the Venn plot and Simpson index results, the MD 5s group exhibited higherα-diversity than the MD 0s and 60s groups. The bacterial community composition of the MD 5s group was similar to that of the MD 0s group, while was different from that of the MD 60s group. The abundances ofPhascolarctobacterium,Blautiaand norank_f_Ruminococcaceae were obviously higher in the MD 0s and MD 5s groups, which might explain their higher production of SCFAs. This was also in line with the Spearman correlation analysis. Results ofinvitroculture ofA.muciniphila,B. acidifaciens,E. coliandB.adolescentiswere in consistency with the results of 16S rDNA sequencing. Fermentation substrates of MD 0s and MD 5s groups promoted the growth of two probiotics -A.muciniphilaandB.adolescentis. Moderately-milled rice (MD 5s) had richer nutrients than white rice (MD 60s) and showed comparative fermentation properties as brown rice (MD 0s).Our results would provide guidance for rice processing industry in terms of the fermentation properties and effects on gut health of rice with different MDs. Fermentation properties of other types of moderately-processed whole grains would also require investigation.How other processing/cooking methods affect the fermentation properties of moderately-processed whole grains would also be an interesting direction.
Conflict of interest
The authors declare no competing financial or non-financial interests.
Acknowledgement
This study was supported by the National Natural Science Foundation of China (32202051), the Shanghai Sailing Program(21YF1431800, 20YF1433400), Shanghai Agriculture Applied Technology Development Program, China (2021-02-08-00-12-F00780) and the National Key R&D Program of China(2022YFF1100104, 2023YFF1103404).
杂志排行

食品科学与人类健康(英文)的其它文章
- Betalains protect various body organs through antioxidant and anti-inf lammatory pathways
- Effects of Maillard reaction and its product AGEs on aging and age-related diseases
- Characterization of physicochemical and immunogenic properties of allergenic proteins altered by food processing: a review
- Polyphenol components in black chokeberry (Aronia melanocarpa)as clinically proven diseases control factors—an overview
- Food-derived protein hydrolysates and peptides: anxiolytic and antidepressant activities, characteristics, and mechanisms
- Recent advances in the study of epitopes, allergens and immunologic cross-reactivity of edible mango