基于HMO方法、FMO理论和DFT计算理解Diels-Alder反应的立体专一性、区域选择性和立体选择性
2024-01-23李唯加柳凯王一翔张志君杨一莹张冬菊
李唯加,柳凯,王一翔,张志君,杨一莹,张冬菊
山东大学化学与化工学院,济南 250100
1 前言
Diels-Alder (D-A)反应是指由共轭双烯烃(双烯体)与取代烯烃(亲双烯体)发生环加成形成取代环己烯的反应,又称双烯合成,或[4+2]环加成。D-A反应是构建C—C键的重要手段,也是现代有机合成里最常用的反应之一,在1928年由德国化学家Otto Diels和他的学生Kurt Alder[1-3]首次发现,二人因此获得1950年的诺贝尔化学奖。
D-A反应是协同反应,新的σ键、π键的形成与旧的π键破裂同时进行,通过一个环状过渡态,转化为产物分子,如式(1)所示。D-A反应呈现丰富的立体化学性质,兼有立体专一性(stereospecificity)、区域选择性(regioselectivity)和立体选择性(stereoselectivity)。这些D-A的选择性是有机化学课程的重要教学内容[4-6],也是结构化学中应用物质结构基本原理理解化学基本概念的具体实例[7-10]。
以有机化学教材[4]中的典型D-A为例,结合休克尔分子轨道方法(Hückel Molecular Orbital,HMO)、前线分子轨道理论(Frontier Molecular Orbital,FMO)和量子化学计算,详细阐述D-A反应的立体专一性、区域选择性和立体选择性。该论文注重化学各基础学科之间的内在关联,强调HMO理论在D-A反应中的应用;启发学生思考D-A反应中蕴含的化学基本原理,引导学生利用分子轨道性质、对称性匹配原理、前线轨道理论等,理解D-A反应的立体专一性、区域选择性和立体选择性;突出计算化学的重要作用,指导学生开展计算化学实验,利用量子化学计算结果加深对D-A反应基本规律的认识,培养学生应用化学基本原理分析和解决实际化学问题的能力。
2 计算方法
以1-甲氧基-1,3-丁二烯和丙烯醛的Diels-Alder反应为例,应用量子化学方法计算了双烯体和亲双烯体的分子结构、前线分子轨道、双烯加成反应的过渡态,研究了反应的热力学和动力学性质。结构优化、振动频率和内禀反应坐标(Intrinsic Reaction Coordinate,IRC)[11],计算使用ωB97X-D泛函[12]和6-311++G(d,p)基组[13],用极化连续介质模型(Polarizable Continuum Model)考虑溶剂化效应[14],使用呋喃做反应溶剂[15]。单点能量计算在MP2(full)[16]/6-311++G(d,p)水平完成,文中给出的能量为吉布斯自由能。全部计算Gaussian 16[17]程序包完成。
3 HMO理论对 和 体系的处理结果
有机化学教材[4]中通常用前线轨道理论和分子轨道对称性守恒原理解释D-A反应的选择性。一般直接给出双烯体和亲双烯体的FMO,包括最高占据分子轨道(HOMO)和最低空轨道(LUMO),这些π分子轨道及其能级应用休克尔分子轨道法求解得到。为简便,这里仅给出HMO求解得到的两个体系的π分子轨道。
对于双烯体:
其中ψ1和ψ2为成键轨道,ψ3和ψ4为反键轨道,ψ2为HOMO,ψ3为LUMO。这些轨道中的组合系数表示对应原子轨道对π分子轨道的贡献,绝对值越大,其贡献越大。将这些分子轨道根据其组合系数的正负及相对大小,将其示于图1a。对于亲双烯体,2个π分子轨道,分别表示为,分别是其HOMO和LUMO。这些轨道图像是理解D-A反应特点和规律的基础。
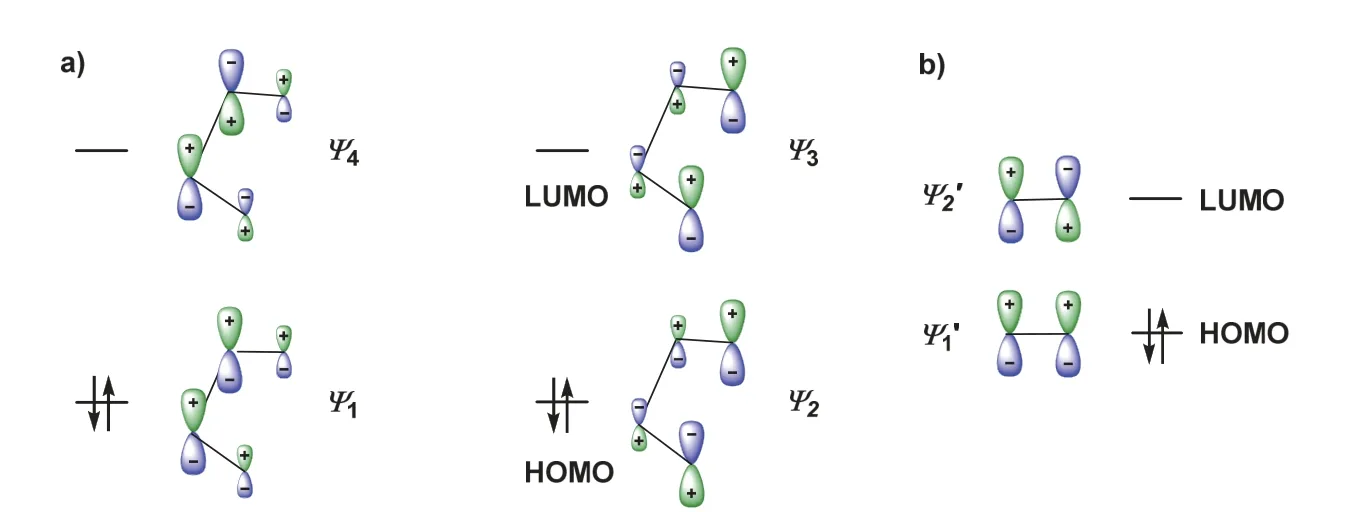
图1 双烯体() (a)与亲双烯体() (b)的π分子轨道
4 Diels-Alder反应的选择性
4.1 立体专一性(Stereospecificity)
D-A反应的专一性是指在加热条件下只能得到顺式加成产物,这可用双烯体和亲双烯体的HOMO和LUMO的对称性予以理解。
从图1容易看出,双烯体的HOMO (ψ2)与亲双烯体的LUMO以及双烯体的LUMO (ψ3)与亲双烯体的HOMO均是对称性匹配的,反应时为了保持轨道有效重叠,需要双烯体两端碳原子发生对旋、双烯体保持顺反关系不变,导致顺式加成产物,因此反应具有立体专一性。
为观察图1中π分子轨道的对称性,我们以图2所示的两个双烯体R1、R3以及亲双烯体R2为例,计算了它们的HOMO、LUMO能级及其等密度面图。通过量子化学计算得到的这些轨道,其对称性与上述用HMO方法处理的结果是一致的。
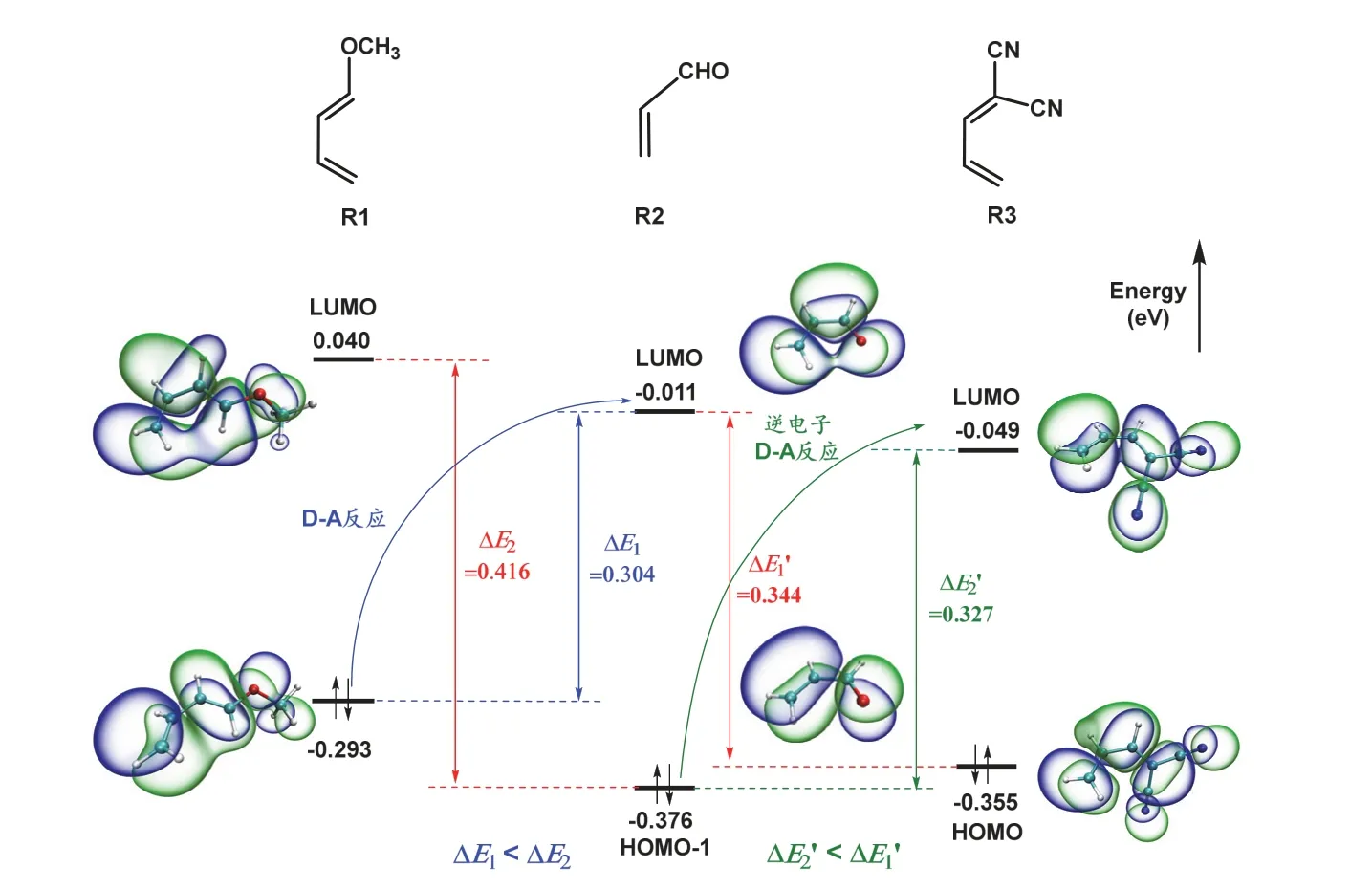
图2 双烯体R1/R3以及亲双烯体R2的HOMO、LUMO能级、等密度面及D-A反应的电子流向
根据反应中电子的流向,可将D-A反应分为传统型和逆电子型。如图2所示,双烯体R1的HOMO能级与亲双烯体R2的LUMO能级之差(ΔΕ1)小于R1的LUMO能级与R2的π电子的最高占据轨道(这里是R2的HOMO-1轨道)能级之差(ΔΕ2)。根据FMO理论,反应时电子将从双烯体R1的HOMO流向亲双烯体R2的LUMO,这样的反应称为传统型D-A反应。当双烯体上有强吸电子基团(如—CN等)时,将降低双烯体π分子轨道能量,使双烯体R3的LUMO能级与亲双烯体R2的HOMO能级差小于双烯体R3的HOMO能级与亲双烯体R2的LUOMO能级的差,反应时电子将从亲双烯体的HOMO流向双烯体的LUMO,这样的反应称为逆电子型D-A反应。
4.2 区域选择性(Regioselectivity)
D-A反应的区域选择性是指双烯体和亲双烯体相对取向不同导致的选择性,当两个反应物上均有取代基时,两个取代基处于邻位的产物是优势产物,而处于间位的产物为副产物。我们以1-甲氧基-1,3-丁二烯和丙烯醛的D-A反应(图3a)为例[4]计算了D-A反应的势能剖面,分别优化了反应物、过渡态和产物的结构,给出了反应的能垒和反应热,主要结果示于图3b。
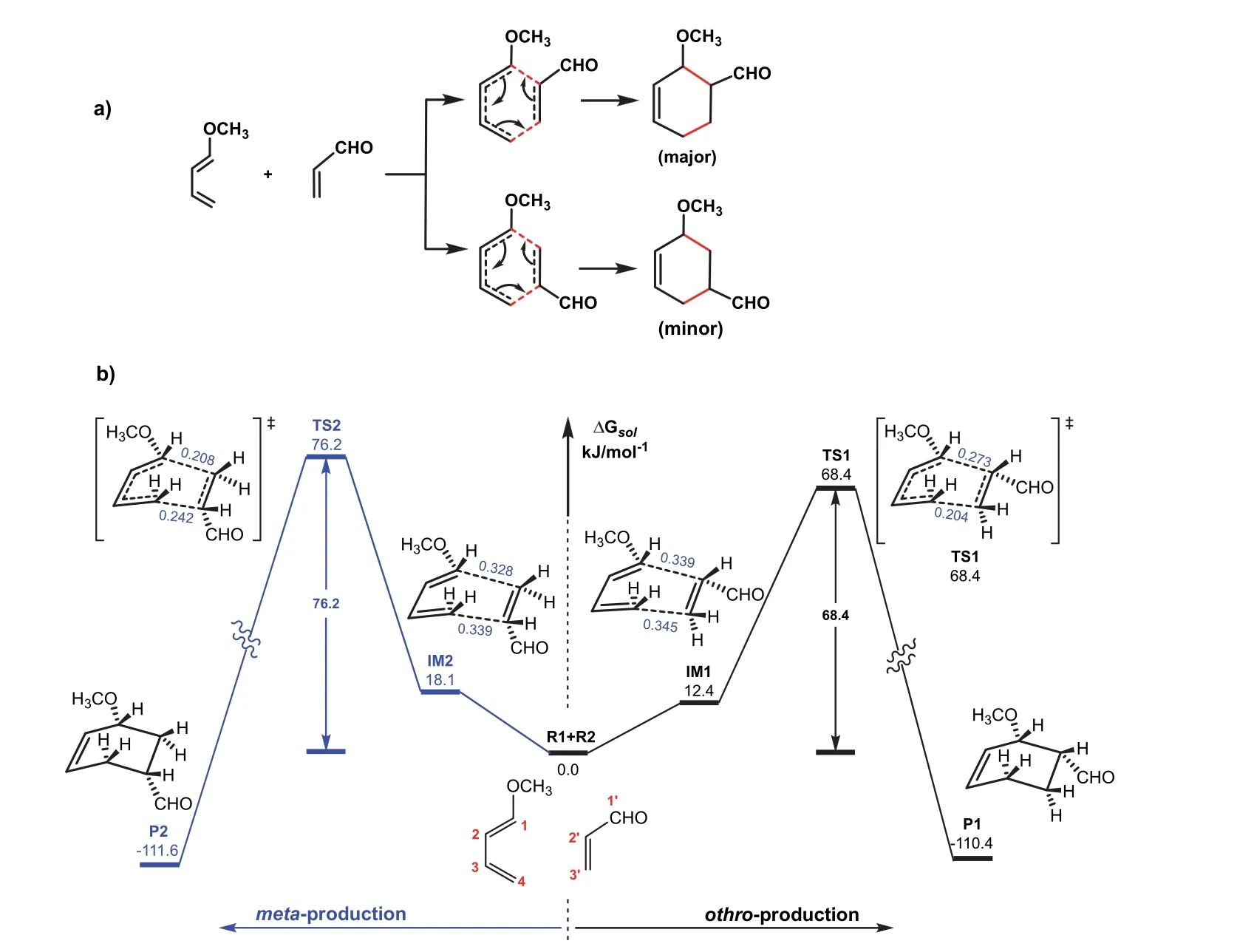
图3 (a) D-A反应的区域选择性;(b) 计算的势能剖面图(邻位(右)与间位(左)产物),过渡态和产物构型均为内型(endo)结构,键长:nm
图中将两底物的能量选为参考点,IM1/IM2、TS1/TS2和P1/P2分别表示双烯体和亲双烯体形成的初始络合物、反应经历的过渡态、邻位和间位产物。为便于比较,这些结构均对应内型结构(定义见4.3节)。从计算的相对能量可以看出,形成邻位和间位产物所需克服的能垒分别为68.4和76.2 kJ·mol-1,反应分别放热110.4和111.6 kJ·mol-1。这些结果表明,形成邻位和间位产物的反应均是不可逆过程,但形成邻位产物经历能量较低的过渡态(TS1),因此邻位产物是主要产物,与实验结果一致。
从过渡态的结构参数可以看出,反应是一个典型的周环反应,通过六元环过渡态一步形成产物,过渡态中旧键断裂与新键形成同时发生。注意,该过程虽协同进行,但两个新σ键的形成是不同步的,如TS1中的结构参数所示,两个正在形成的C-C距离分别为0.204和0.273 nm。这样,1-甲氧基-1,3-丁二烯和丙烯醛的反应是一个协同不同步的基元过程。图4给出的IRC计算结果进一步证明了该反应协同不同步特征。
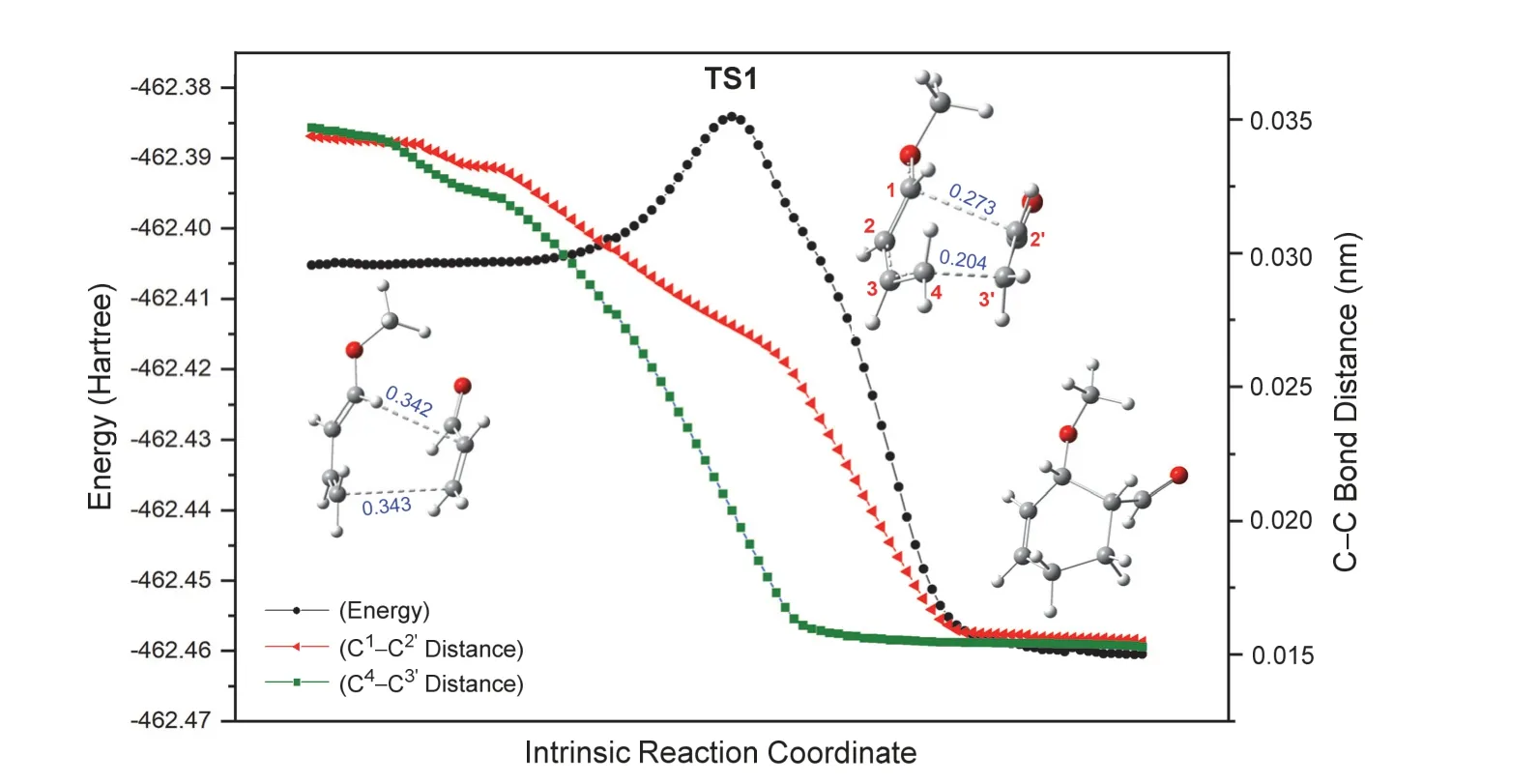
图4 TS1的IRC计算结果
D-A反应是[4+2]环加成反应,反应中的主要轨道相互作用双烯体和亲双烯体p轨道间的重组过程,如图5a中黑色虚线所示。与间位产物的过渡态TS2相比,邻位产物TS1较高的稳定性通常认为是由于该结构中存在较有效的次级轨道相互作用,用红色虚线示于图5b。为可视化这些轨道相互作用,我们比较了两个过渡态的HOMO (图5b)以及两个过渡态中双烯体和亲双烯体间的相互作用(图5c、5d)。如图5b所示,在TS1的HOMO中可以明显看到双烯体(R1)甲氧基与亲双烯体(R2)羰基之间的p-π相互作用;而在TS2中,空间因素和轨道对称性均不利于这种次级轨道相互作用。图5c和5d分别表示对两个过渡态进行IRI (Interaction Region Indicator)分析[18]和IGMH (Independent Gradient Model based on Hirshfeld partition)分析[19]的结果,两种相互作用分析方法进一步证实了TS1中明显的p-π轨道重叠,即甲氧基O的孤对p轨道与羰基C—O双键π成键轨道之间有利的相互作用。因此,D-A反应主要通过TS1形成邻位产物。
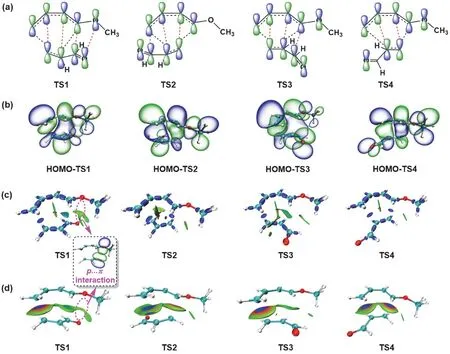
图5 (a) 轨道相互作用(黑色虚线)及次级轨道相互作用(红色虚线)示意图;(b) HOMO等密度面;(c) IRI等密度面;(d) IGMH等密度面
4.3 立体选择性(Stereoselectivity)
D-A反应的立体选择性是指当亲双烯体为不对称结构时,如图6a所示,优先形成内型(endo)加成产物,即亲双烯体上共轭的不饱和取代基于双烯体中新形成的双键位于连接平面同侧的产物,而外型(exo)产物(取代基与双键位于连接平面异侧)为次要产物。
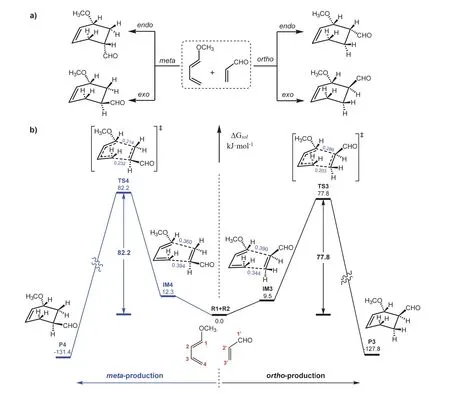
图6 (a) D-A反应的区域选择性(邻位 vs. 间位)和立体选择性(内型 vs. 外型);(b) 计算的势能剖面图:邻位(右)与间位(左)产物,过渡态和产物构型均为外型结构,键长:nm
如上所述,在分析邻、对位区域选择性时,选取的均为内型结构。为了进一步理解D-A反应的立体选择性,比较内、外型产物对应过渡态的能量,我们进一步使用外型结构计算了D-A反应的势能剖面图,结果示于图6b。图中TS3和TS4分别表示形成exo型邻位和间位产物的过渡态。可以看出,与内型结构类似(图3b),也是邻位过渡态能量更低(77.8vs. 82.2 kJ·mol-1)。另一方面,比较图6b和图3b,内型过渡态结构(TS1和TS2)均比对应的外型过渡态结构(TS3和TS4)更稳定,因此形成的产物主要为内型产物。TS3和TS4的HOMO及对其进行IRI和IGMH分析的结果一并在图5中给出。显然,由于空间因素,次级轨道相互作用较弱,特别是甲氧基与羰基之间缺少p-π相互作用,致使两个外型过渡态的能量较高。这些结果与教材[4]中定性描述完全一致。
5 结语
以1-甲氧基-1,3-丁二烯和丙烯醛的Diels-Alder反应为例,设计了一个研究型计算化学实验。通过量子化学计算了势能面上稳定点的结构和性质,给出了反应的过渡态结构以及热力学和动力学数据,结合休克尔分子轨道理论和前线轨道理论合理解释了反应的立体专一性、区域选择性和立体选择性。研究过程有助于培养学生从事科研工作的兴趣以及分析问题和解决问题的能力,研究手段有助于加深学生对计算化学的理解,研究结果有助于学生全面掌握D-A反应的特点和规律。
