全氟立方烷的笼状立体电子效应
2024-01-23李懿伦袁耀锋叶克印
李懿伦,袁耀锋,叶克印
福州大学化学学院,福州 350108
1 前言
立体电子效应是有机化学理论的重要组成部分[1],作为超共轭效应的延伸,立体电子效应可以解释有机化学中很多的“反常”现象,包括1,2-二氟乙烷的邻位交叉效应(Gauche Effect)[2,3]、糖化学中的异头碳效应(Anomeric Effect)[4,5]、米氏酸的反常酸性[6],甚至近期有计算化学研究表明,乙烷邻位交叉构象为优势构象与C—H键的超共轭效应有着密切的关系[7-9]。
立体电子效应本质是分子轨道间的电子给体-受体相互作用。在基于空间位阻的构象理论中,分子中两个会形成较大排斥的取代基总是倾向处于距离最远的位置,形成对位交叉的优势构象。而在1,2-二氟乙烷中,氟原子的强电负性(3.98)使得C—F键被强烈极化,进而导致C—F键的反键轨道能量显著地下降[10],使得C—F键的反键轨道成为了很好的电子受体,C—H键成键轨道对其的超共轭效应显著增强,最终促进了邻位交叉构象的稳定性(图1)。
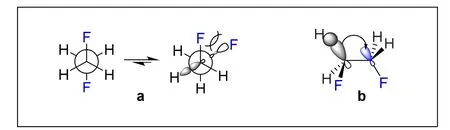
图1 邻位交叉效应
事实上,C—F键的反键轨道作为电子受体的性质也引起了许多理论化学家的关注,许多计算化学工作也对相应的碳-氟骨架的电子接受能力进行了研究。本文将基于计算化学的相关工作与最新的对全氟立方烷合成及性质研究的实验工作,对全氟立方烷及C—F反键构成的电子受体进行回顾和讨论。
2. 全氟立方烷的电子受体性质
2.1 C—F电子受体的理论预测
根据分子的结构可知,环状或者笼状的C—F反键轨道是指向分子中央的,如果这个环或笼的大小合适,这些反键轨道可以在分子中央部分重叠,使得这类分子可能成为良好的电子受体。2008年,Karl K. Irikura课题组对C—F化合物的电子接受能力进行了总结[11],作者通过整理和计算不同碳氟化合物的电子亲合能,来探究环状或笼状碳氟化合物作为电子受体的能力。电子亲和能EA (Electron Affinity)是指中性分子或原子接受一个电子形成阴离子所释放的能量,因此电子亲合能的正值越大则说明分子或原子的电子接受能力越强。例如,电负性最高的氟元素的电子亲合能为3.40 eV,而电负性较小的碳元素的电子亲合能为1.26 eV。
通过对环状碳氟化合物分子能量的计算,作者发现从三元环到六元环(图2),环状碳氟化合物的电子亲合能均为正值,说明其具有一定的电子接受能力从而形成相对稳定的自由基阴离子形式。全氟环丙烷(图2a)的电子亲合能为0.74 eV,随着环的扩大,全氟环丁烷(图2b)和全氟环戊烷(图2c)的电子亲合能显著增加,均为1.11 eV,这可能是因为环扩大后使得各个C—F键的反键轨道的重叠程度更好,电子接受能力明显提高。对于六元环的全氟环己烷(图2d),其电子亲合能为0.77 eV,明显低于四元环和五元环的体系,这可能也与分子的几何构型和C—F键反键轨道的重叠程度密切相关。
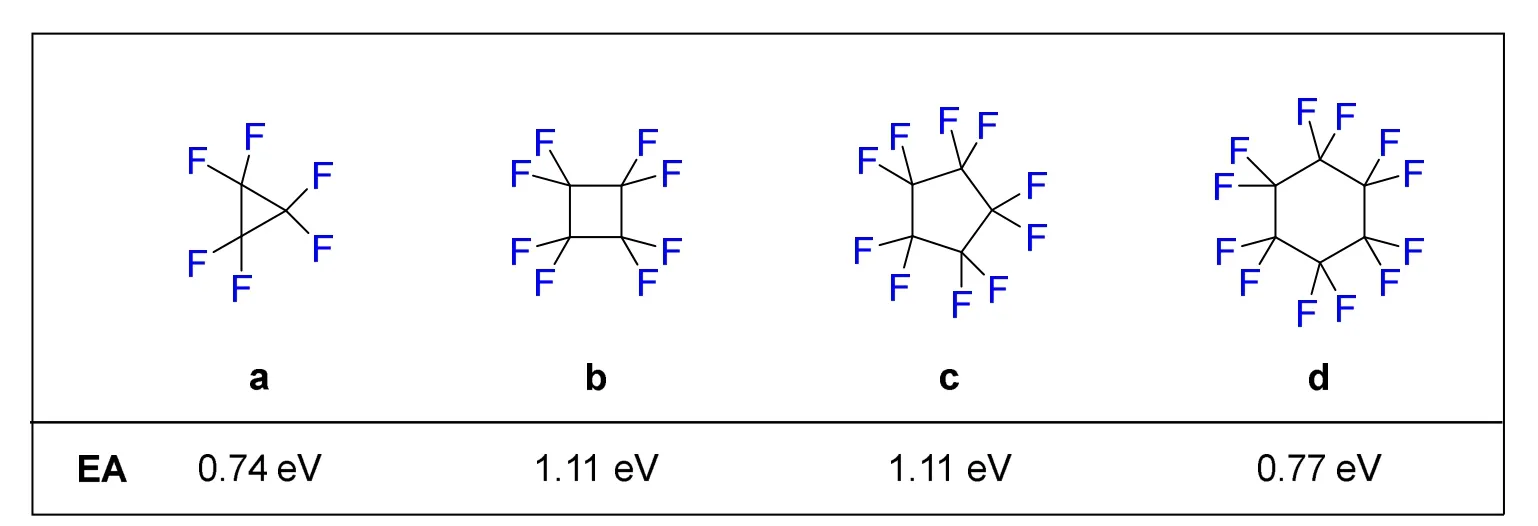
图2 环状碳氟化合物
同样,作者也分别对立方烷(图3a)与全氟立方烷(图3b)的电子亲合能进行了计算。立方烷的电子亲合能是-1.14 eV,而全氟取代的立方烷的电子亲合能约为1.30 eV,这表明全氟立方烷具有非常优异的电子接受能力。从分子轨道可以看出,每一个缺电子C—F键的反键轨道均指向全氟立方烷分子的中心位置,八根C—F键的反键轨道可以在分子中心位置部分重叠,形成能量更低的分子轨道,从而更容易接受电子。
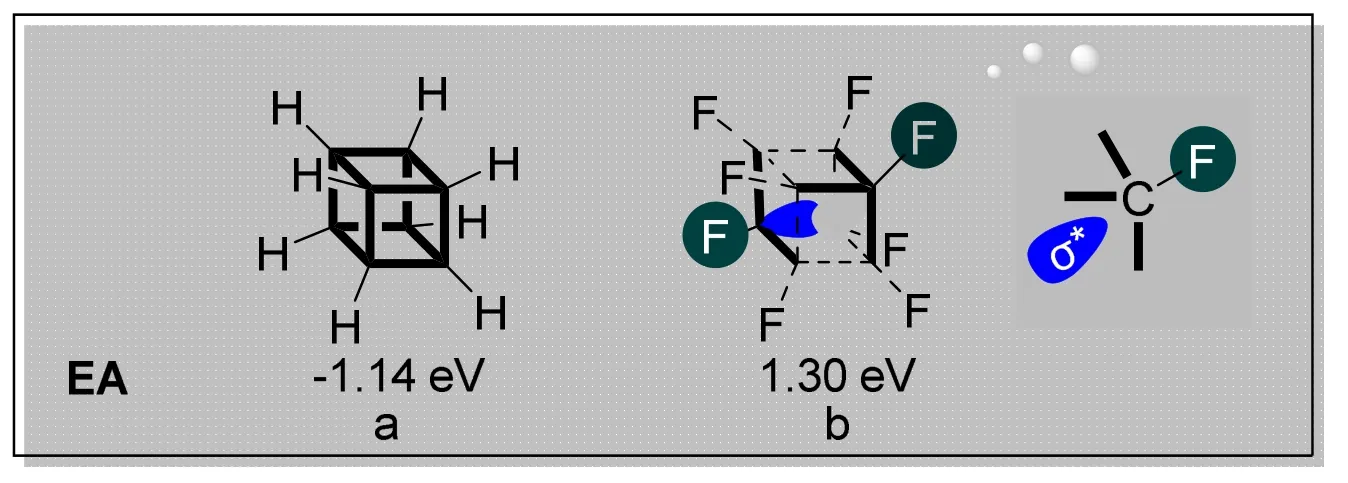
图3 立方烷和全氟立方烷
2.2 全氟立方烷的合成
事实上,早在2004年就已经有理论化学工作预测了全氟立方烷的存在和相应的理化性质[12],但由于立方烷骨架的特殊性,直到2022年,Takashi Okazoe课题组才第一次完成了全氟立方烷的合成工作(图4)[13]。为了避免立方烷骨架被剧烈的氟代条件破坏,作者选择了强吸电子基取代的立方烷1作为底物,经过氟化后得到相应的七氟代立方烷化合物2。化合物2未经分离,直接在碱性条件下与苄醇进行酯交换得到化合物3。化合物3经分离纯化后进行水解脱羧反应得到化合物4,最后在强碱的作用下与N-氟代双苯磺酰胺(NFSI)发生氟代反应得到最终的全氟立方烷产物5。
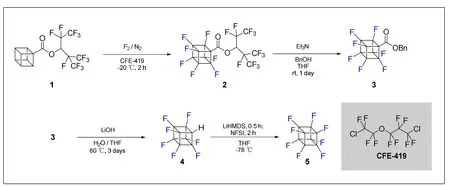
图4 全氟立方烷的合成
2.3 全氟立方烷的结构表征与性质研究
对全氟立方烷产物进行X射线单晶衍射表征,发现全氟立方烷的12根C—C键键长均为1.570 Å(1 Å = 0.1 nm),这同之前通过高精度密度泛函理论(DFT)计算得到的结果基本一致[14]。在立方烷C8H8中,C—C键的键长为1.572 Å,这同全氟立方烷中的键长也十分接近。如果仅从空间位阻角度来看,这样的结果似乎是不合理的,因为引入了8个氟原子间存在着一定的排斥作用,会导致C—C键键长因为相互排斥而增长。然而,由于氟具有很强的电负性,根据本特规则(Bent’s rule)[15],C—F键的杂化轨道中具有更多的p成分,相应的C—C键的杂化轨道中会具有更多的s成分,s成分的增加会导致键长缩短。因此,导致键长缩短的强电负性与导致键长增加的空间排斥作用相互抵消,最终使得引入氟原子并没有对立方烷骨架的C—C键键长产生明显影响。
有趣的是,作者发现在单晶结构中,存在一种弱相互作用,即全氟立方烷分子的一个氟原子会指向邻近的另一个分子的四元环平面中心(图5a),这也从另一个角度揭示了全氟立方烷分子作为电子受体的能力。有理论化学工作曾对全氟立方烷做过分子表面静电势的分析(MPE Analysis)[16],发现全氟立方烷的每一个四元环平面都具有一个σ空穴(σ-hole),即分子静电势能面上正电荷集中分布的位置,可以同作为电子给体的Lewis碱相互作用。以氰离子为例,全氟立方烷同氰离子的结合能为-0.79 eV (图5b,负值表示该结合过程释放能量,是一个热力学有利的过程)。
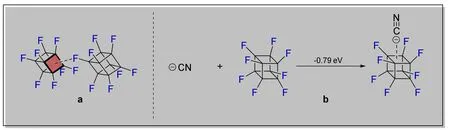
图5 全氟立方烷中的弱相互作用
作者通过密度泛函理论,对全氟立方烷5及其单氢4、双氢6取代的化合物进行了进一步的理论计算(图6)。计算结果表明,随着氟取代数目的增多,化合物的最高占有轨道(LUMO)的能量逐渐降低,双氢代6、单氢代4、全氟立方烷5的LUMO能量分别为-1.3、-2.2、-2.8 eV,这也证明了全氟立方烷独特的电子接受能力。同理论计算结果相符,紫外-可见吸收光谱实验表明,全氟立方烷5在三个化合物中具有最长的吸收波长,并且随着氟取代数目的减少,吸收波长逐渐蓝移。
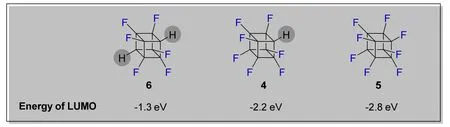
图6 全氟立方烷及其氢取代衍生物的LUMO能量
事实上,配位原子对平面上σ空穴配位的例子在无机化学中也有报道。例如,Boudjouk课题组合成了对甲基苯甲腈分子和卤素离子等一系列配位分子对全卤代环己硅烷配位的三明治夹心型化合物(图7),包括Si6Br12·2(p-CH3C6H4CN) 7[17]与(n-Bu4N)2(Si6Cl12I2) 8[18],表明具有σ空穴的化合物作为电子受体具有一定的广谱适用性。
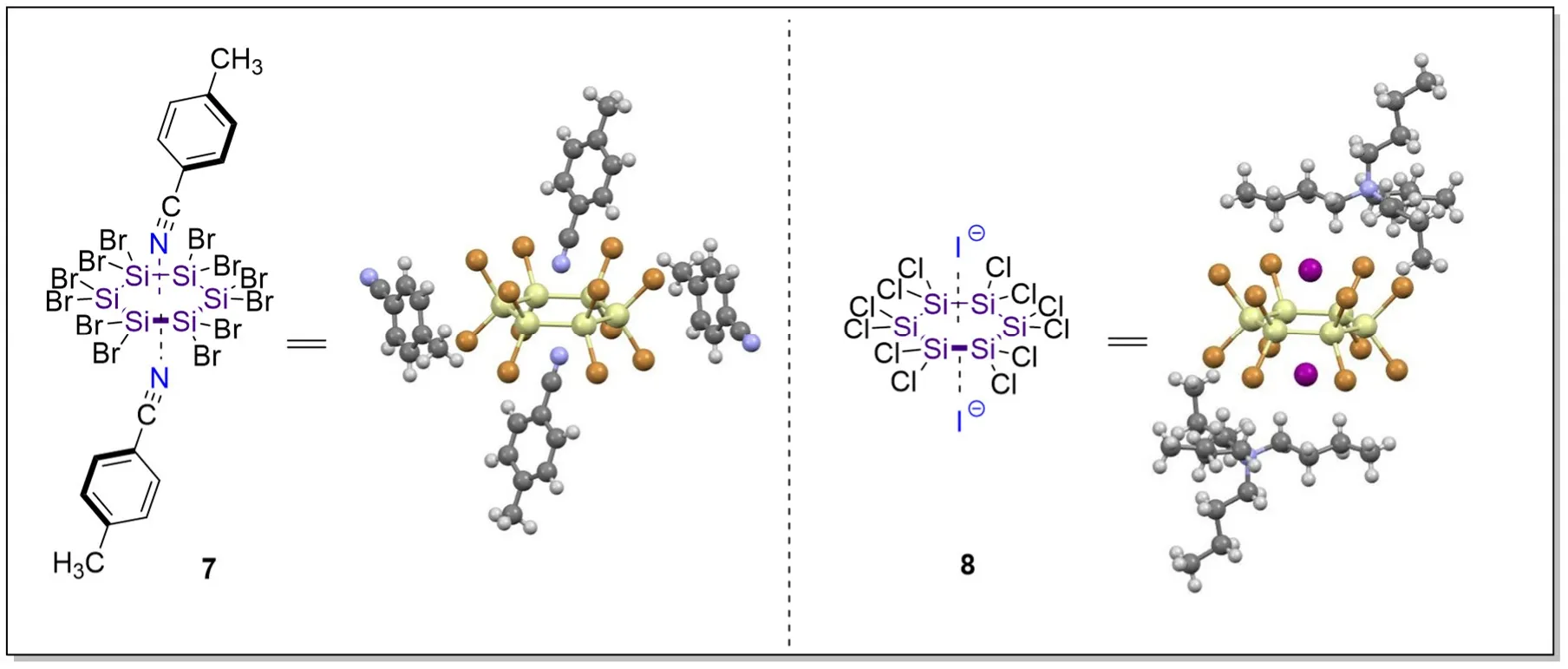
图7 其他具有σ空穴的化合物作为电子受体
3 结语
作为基础有机化学的重要组成部分,立体电子效应可以解释许多有机化学中独特的现象。通过化学家们多年的研究,从最初的理论预测到全氟立方烷的成功合成,越来越多的C—F键反键轨道作为电子受体的例子被研究和报道。随着理论化学与合成化学的进一步发展,立体电子效应必将得到进一步的发展和应用。
