金属卡宾模板导向的多咪唑鎓盐大环合成
2024-01-20王以寿王尧宇韩英锋
王以寿 白 莎 王尧宇 韩英锋
(1西北大学化学与材料科学学院,西安 710127)
(2商洛学院化学工程与现代材料学院,商洛 726000)
0 Introduction
Metal-directed self-assembly stands as a pivotal synthetic strategy in the construction of supramolecular structures, consistently yielding valuable metallosupramolecular entities of diverse shapes and sizes[1-5].Traditionally,structures of this kind have been predominantly fashioned from Werner - type complexes, wherein polydentate ligands featuring N or O donor atoms are coordinated to metal centers[6-11].Moreover, the integration of extended organometallic fragments such as M—CNHC(NHC=N-heterocyclic carbene) building blocks has facilitated the development of innovative assemblies[12-15].Capitalizing on the exceptional stability intrinsic to selected M—CNHCbonds, a spectrum of organometallic molecular assemblies has been meticulously designed and explored, employing a variety of polydentate NHC ligands[16-21].Owing to their captivating structures and remarkable properties in the realms of sensing, catalysis, and optics, these assemblies find widespread applications in host - guest chemistry,template-controlled photoreactions, and selective catalytic transformations[22-30].
Macrocycles, especially those incorporating extensive heterocyclic structures with diverse ring sizes and functionalities, have discovered intriguing applications in materials, pharmaceuticals, and supramolecular science[31-35].Their synthesis has frequently been approached through ring - closing metathesis (RCM)reactions, standing as a pivotal step in the synthetic process.In historical context, the narrative of nonstereoselective macrocyclic RCM reactions commenced in 1980[36-37].In the late 1990s,the RCM-based syntheses of epothilone and the 18-memberedα,βunsaturated macrolide aspicilin were deemed as pioneering total syntheses of natural macrocyclic compounds utilizing the RCM strategy[38-40].Subsequent to these milestones, the development of efficient catalysts, exemplified by ruthenium-based Grubbs′ complexes and molybdenum Schrock′s catalysts, has firmly established RCM as one of the most extensively employed methodologies in the design and synthesis of macrocycles[41-43].
Despite notable achievements in the field, there exists substantial potential for the expansion of the functional macrocycle class.Macrocyclization reactions often contend with undesired intermolecular reactions[44-45].Conventional synthetic methodologies for macrocycles prove impractical and inefficient, frequently resulting in diminished yields and the occurrence of unwanted side reactions.This study introduces the construction of ordered structures derived from poly-NHC precursors via template-directed RCM.Our approach capitalizes on metal-directed self-assembly in conjunction with a suitable template to facilitate the RCM of linear dienes.This strategic amalgamation enables the promotion of the desired dinuclear silver (Ⅰ)tetracarbene complex, ultimately leading to the formation of the corresponding covalent organic macrocycle upon removing silver ions.
1 Experimental
1.1 Materials and physical measurements
1H and13C{1H} spectra were acquired on a Bruker AVANCE Ⅲ400 spectrometer.Chemical shifts (δ) are reported in parts per million relative to tetramethylsilane, with the residual protonated solvent serving as an internal standard.Mass spectra were generated using a Bruker microTOF-Q Ⅱ mass spectrometer (Bruker Daltonics Corp., USA) employing the electrospray ionization (ESI) mode.Unless explicitly mentioned, all reagents were commercially sourced and employed without additional purification.
1.2 Synthesis of the ligands and macrocycles
1.2.1 Synthesis of H2-A1(BF4)2
A Schlenk flask was charged with 4-(imidazole-1-yl)phenol(0.705 g,4.4 mmol)and 1,4-bis(bromomethyl)benzene (0.528 g, 2.0 mmol).To this mixture, DMF was added (2 mL), and the reaction mixture was heated to 110 ℃for 12 h.After cooling the reaction mixture to ambient temperature, 30 mL ethyl acetate was added,and a white compound precipitated.The solid was isolated by filtration, washed with ethyl acetate, and driedinvacuoto give a white solid.Yield: 1.138 g (1.95 mmol, 97%).The obtained solid (0.292 g, 0.5 mmol)was transferred to a bottle containing 5-bromo-1-pentene (0.6 mL, 5 mmol), NaOH (0.060 g, 1.5 mmol) and 40 mL ethanol.The mixture was stirred for 48 h under reflux.Then, remove the solvent and the solid was washed with water.The solid was isolated by filtration and transferred to a bottle containing 10 mL methanol;a solution of NH4BF4(0.210 g, 2.0 mmol) in methanol(20 mL) was added.The resulting mixture was stirred at ambient temperature for 6 h.Then, the solvent was removed, and the solid was washed with water and driedinvacuo.Yield: 0.312 g (0.43 mmol, 85%, two steps).1H NMR (400 MHz, DMSO-d6):δ=9.96 (s, 2H,H1),8.25(s,2H,imidazolium-H),8.01(s,2H,imidazolium-H),7.70(d,J=8.8 Hz,4H,Ar-H),7.61(s,4H,Ar-H), 7.18 (d,J=8.8 Hz, 4H, Ar-H), 5.92-5.82 (m, 2H,H14),5.52(s,4H,H4),5.08-4.98(m,4H,H15),4.06(t,J=6.4 Hz,4H,H11),2.20(q,J=6.8 Hz,4H,H13),1.83(p,J=6.6 Hz, 4H, H12).13C{1H} NMR (100 MHz,DMSO-d6):δ=159.4,137.8,135.3,135.1,129.1,127.7,123.5, 123.0, 121.9, 115.6, 115.3, 67.4, 51.8, 29.5,27.7.ESI-MS (positive ions):m/z=280.157 0 (Calcd.for[H2-A1]2+280.141 3).
1.2.2 Synthesis of H2-B1(BF4)2
1.2.3 Synthesis of[Ag2(A1)2](BF4)2
A sample of H2-A1(BF4)2(73 mg, 0.1 mmol) was dissolved in 25 mL of CH3CN, and to this solution Ag2O (46 mg, 0.2 mmol) was added.The resulting suspension was heated to 70 ℃for 24 h without light.After cooling to ambient temperature, the obtained suspension was filtered through a pad of Celite to give a clear solution.The filtrate was concentrated to 3 mL,and diethyl ether (30 mL) was added, leading to the precipitation of [Ag2(A1)2](BF4)2as a yellow solid.The solid was filtrated,washed with diethyl ether,and driedinvacuo.Yield: 62 mg (0.041 mmol, 82%).1H NMR(400 MHz, DMSO-d6):δ=7.81 (s, 4H, Ar-H), 7.73 (s,4H, Ar-H), 7.48 (d,J=8.4 Hz, 8H, Ar-H), 7.13 (s, 8H,Ar-H), 6.92 (d,J=8.4 Hz, 8H, Ar-H), 5.97-5.74 (m,4H, H14), 5.29 (s, 8H, H4), 5.14-4.88 (m, 8H, H15),3.99 (s, 8H, H11), 2.19 (s, 8H, H13), 1.83 (s, 8H,H12).13C{1H} NMR (100 MHz, DMSO-d6):δ=158.5,138.8,136.7,132.6,127.7,125.1,123.1,122.9,115.2,115,67.2,53.9,29.5,27.7.ESI-MS(positive ions):m/z=666.196 6(Calcd.for[Ag2(A1)2]2+666.204 3).
1.2.4 Synthesis of[Ag2(B1)2](BF4)2
A sample of H2-B1(BF4)2(0.810 g, 1.0 mmol) was dissolved in 40 mL of CH3CN, and to this solution Ag2O(0.695 g,3.0 mmol)was added.The resulting suspension was heated to 70 ℃for 24 h without light.After cooling to ambient temperature, the obtained suspension was filtered through a pad of Celite to give a clear solution.The filtrate was concentrated to 3 mL,and diethyl ether (30 mL) was added, leading to the precipitation of [Ag2(B1)2](BF4)2as a yellow solid.The solid was filtrated,washed with diethyl ether,and driedinvacuo.Yield: 0.819 g (0.49 mmol, 98%).1H NMR(400 MHz, DMSO-d6):δ=7.83 (s, 4H, H2), 7.80 (s, 4H,H3), 7.55 (d,J=8.6 Hz, 8H, H10), 7.35 (d,J=7.4 Hz,8H, H7), 7.18 (d,J=7.4 Hz, 8H, H6), 6.95 (d,J=8.6 Hz, 8H, H11), 6.01-5.71 (m, 4H, H16), 5.45 (s, 8H,H4), 5.15-4.90 (m, 8H, H17), 4.01 (t,J=6.1 Hz, 8H,H13), 2.22 (q,J=6.5 Hz, 8H, H15), 1.85 (p,J=6.2 Hz,8H, H14).13C{1H} NMR (100 MHz, DMSO-d6):δ=178.1 (C1),158.5 (C12),138.8 (C8),137.8 (C16),136.2(C5), 132.5 (C9), 127.7 (C6), 126.8 (C7), 125.1 (C10),123.2 (C2), 123.2 (C3), 115.3 (C17), 115.1 (C11), 67.2(C13),54.1(C4),29.6(C15),27.8(C14).ESI-MS(positive ions):m/z=1 571.445 1 (Calcd.for [Ag2(B1)2(BF4)]+1 571.476 1),m/z=742.255 2 (Calcd.for [Ag2(B1)2]2+742.235 8).
1.2.5 Synthesis of[Ag2(A2)](BF4)2
A sample of [Ag2(A1)2](BF4)2(151 mg, 0.1 mmol)and Grubbs′2nd catalyst (42 mg,0.05 mmol)were dissolved in dry CH2Cl2(80 mL) under a nitrogen atmosphere.The reaction mixture was stirred at 40 ℃for 12 h.After the reaction was cooled to room temperature,10 mL acetonitrile was added to the mixture, and the obtained mixture was filtered to give a clear solution.The filtrate was concentrated to 3 mL, and ethyl acetate (30 mL) was added, leading to the precipitation of[Ag2(A2)](BF4)2as a gray solid.The solid was collected by filtration, washed with ethyl acetate, and driedin vacuo.Yield: 0.119 g (0.085 mmol, 85%).[Ag2(A2)](BF4)2was used for subsequent reactions without further purification.
1.2.6 Synthesis of[Ag2(B2)](BF4)2
A sample of [Ag2(B1)2](BF4)2(100 mg, 0.06 mmol)and Grubbs′2nd catalyst (10 mg,0.01 mmol)were dissolved in dry CH2Cl2(80 mL) under a nitrogen atmosphere.The reaction mixture was stirred at 40 ℃for 12 h.After the reaction was cooled to room temperature,10 mL acetonitrile was added to the mixture, and the obtained mixture was filtered to give a clear solution.The filtrate was concentrated to 3 mL, and diethyl ether(30 mL)was added,leading to the precipitation of[Ag2(B2)](BF4)2as a gray solid.The solid was filtrated,washed with diethyl ether, and driedinvacuo.Yield:79 mg(0.049 mmol,82%).1H NMR(400 MHz,DMSOd6):δ=7.83 (s, 4H, H3), 7.77 (s, 4H, H2), 7.57 (d,J=8.8 Hz,8H,H10),7.27(d,J=8.0 Hz,8H,H7),7.10(d,J=8.0 Hz, 8H, H6), 6.92 (d,J=8.8 Hz, 8H, H11), 5.62-5.35 (m, 8H, H16, H4), 4.06-3.90 (m, 8H, H13), 2.27-2.13 (m, 8H, H15), 1.96-1.70 (m, 8H, H14).13C{1H}NMR(100 MHz,DMSO-d6):δ=176.9(C1),158.6(C12),138.6 (C8), 136.0 (C5), 132.5 (C9), 130.2 (C16), 127.3(C6), 126.6 (C7), 125.0 (C10), 123.3 (C2, C3), 115.1(C11), 67.0 (C13), 54.1 (C4), 28.5 (C15), 27.6 (C14).ESI-MS (positive ions):m/z=1 515.391 9 (Calcd.for[Ag2(B2)(BF4)]+1 515.413 4),m/z=714.205 6 (Calcd.for[Ag2(B2)]2+714.204 5).
高邮湖地处苏皖交界,北与洪泽湖水系相连接,南与长江水系相连通,东临江苏高邮,西接安徽天长,跨安徽省天长市和江苏省高邮市、宝应县、金湖县。高邮湖湖区主属江苏省,是江苏省第三大湖,水域总面积为760.67 km2,在高邮市境内水域面积392.82 km2,占高邮湖总水域面积的55.32%。高邮湖属浅水型湖泊,由古泻湖经长期淤积和人类活动影响而成。
1.2.7 Synthesis of H4-A2(BF4)4
[Ag2(A2)] (BF4)2(0.060 g, 0.041 mmol)was dissolved in CH3CN (5 mL).To this was added NH4Cl(0.007 g, 0.124 mmol) in methanol (5 mL).White solid AgCl precipitated immediately.The resulting suspension was filtered through Celite to obtain a clear solution.The solvent was removed to give a gray solid.The solid was washed with water and then suspended in CH3CN (2 mL), and a solution of NH4BF4(0.013 g,0.124 mmol) in methanol (2 mL) was added.The mixture was stirred at ambient temperature for 12 h.The solvent was removed, and the solid was washed with water and driedinvacuo.Yield: 44 mg (0.031 mmol,76%).1H NMR (400 MHz, DMSO-d6):δ=9.81 (s, 4H,H1),8.21(s,4H,imidazolium-H),8.00(s,4H,imidazolium-H), 7.70-7.61 (m, 8H, Ar-H), 7.57 (s, 8H, Ar-H),7.22-7.13 (m,8H,Ar-H),5.48 (s,12H,H4,H14),4.03(s, 8H, H11), 2.14 (s, 8H, 13), 1.78 (s, 8H, H12).13C{1H} NMR (100 MHz, DMSO -d6):δ=159.4, 135.2,129.8, 129.1, 127.6, 123.5, 123.0, 121.9, 115.6, 67.4,51.9,28.2,23.0.ESI-MS(positive ions):m/z=266.147 8(Calcd.for[H4-A2]4+266.141 4).
1.2.8 Synthesis of H4-B2(BF4)4
A sample of[Ag2(B2)](BF4)2(0.069 g,0.043 mmol)was dissolved in CH3CN (10 mL).To this was added NH4Cl (0.007 g, 0.13 mmol) in methanol (5 mL).A white solid (AgCl) precipitated immediately.The reaction mixture was stirred for 6 h.The resulting suspension was filtered through a pad of Celite to obtain a clear solution.The solvent was removed to give a gray solid.The gray solid was dissolved in methanol (10 mL), and a solution of NH4BF4(0.014 g, 0.13 mmol) in methanol (10 mL) was added.The mixture was stirred at ambient temperature for 12 h.The solvent was removed, and the solid was washed with water and driedinvacuo.Yield: 0.052 g (0.033 mmol, 77%).1H NMR (400 MHz, DMSO-d6):δ=9.86 (s, 4H, H1), 8.23(s, 4H, H2), 8.03 (s, 4H, H3), 7.84-7.71 (m, 8H, H7),7.71-7.64 (m, 8H, H10), 7.64-7.55 (m, 8H, H6), 7.25-7.11(m,8H,H11),5.64-5.43(m,12H,H16,H4),4.20-3.78 (m, 8H, H13), 2.27-2.08 (m, 8H, H15), 1.88-1.66(m, 8H, H14).13C{1H} NMR (100 MHz, DMSO-d6):δ=159.4 (C12), 139.8 (C8), 135.2 (C1), 134.0 (C5), 129.8(C16), 129.2 (C6), 127.7 (C9), 127.3 (C7), 123.6 (C10),123.1 (C3), 121.9 (C2), 115.6 (C11), 67.3 (C13), 52.0(C4), 28.3 (C15), 26.6 (C14).ESI-MS (positive ions):m/z=1 477.616 5(calcd.for[H4-B2(BF4)3]+1 477.641 7),695.317 2 (Calcd.for [H4- B2(BF4)2]2+695.318 6),434.548 7 (Calcd.for [H4- B2(BF4)]3+434.544 3),304.161 1(Calcd.for[H4-B2]4+304.157 0).
2 Results and discussion
In contrast to the numerous molecular macrocycles constructed through metal coordination and hydrogen bonding,the synthesis of covalent organic macrocycles poses greater challenges due to their heightened thermal and chemical stability[46-47].Consequently, template-directed synthesis has been acknowledged as a rational strategy for the intricate construction of such macrocycles.Here, we introduce a rectangular metallacycle assembled by two bidentate ligands and two metal (Ⅰ)ions.Two terminal olefins were incorporated into the ligands,positioning them in proximity.With an appropriate linker, these positions were designed for cross - linking neighboring ligands through olefin metathesis reactions.Given its high thermal stability and resistance to Grubbs′ 2nd catalyst, we selected an Ag(Ⅰ)metallacycle.Our approach showcases the highly efficient synthesis of a covalent organic macrocycle,commencing with H2-L1(BF4)2,through a four-step process (Scheme 1): (1) the design and preparation of the ligand; (2) NHC-directed self-assembly; (3) ring-closing metathesis (RCM) reaction between the adjacent olefins on the Ag(I) metallacycle; (4) removal of the Ag(Ⅰ)ions.
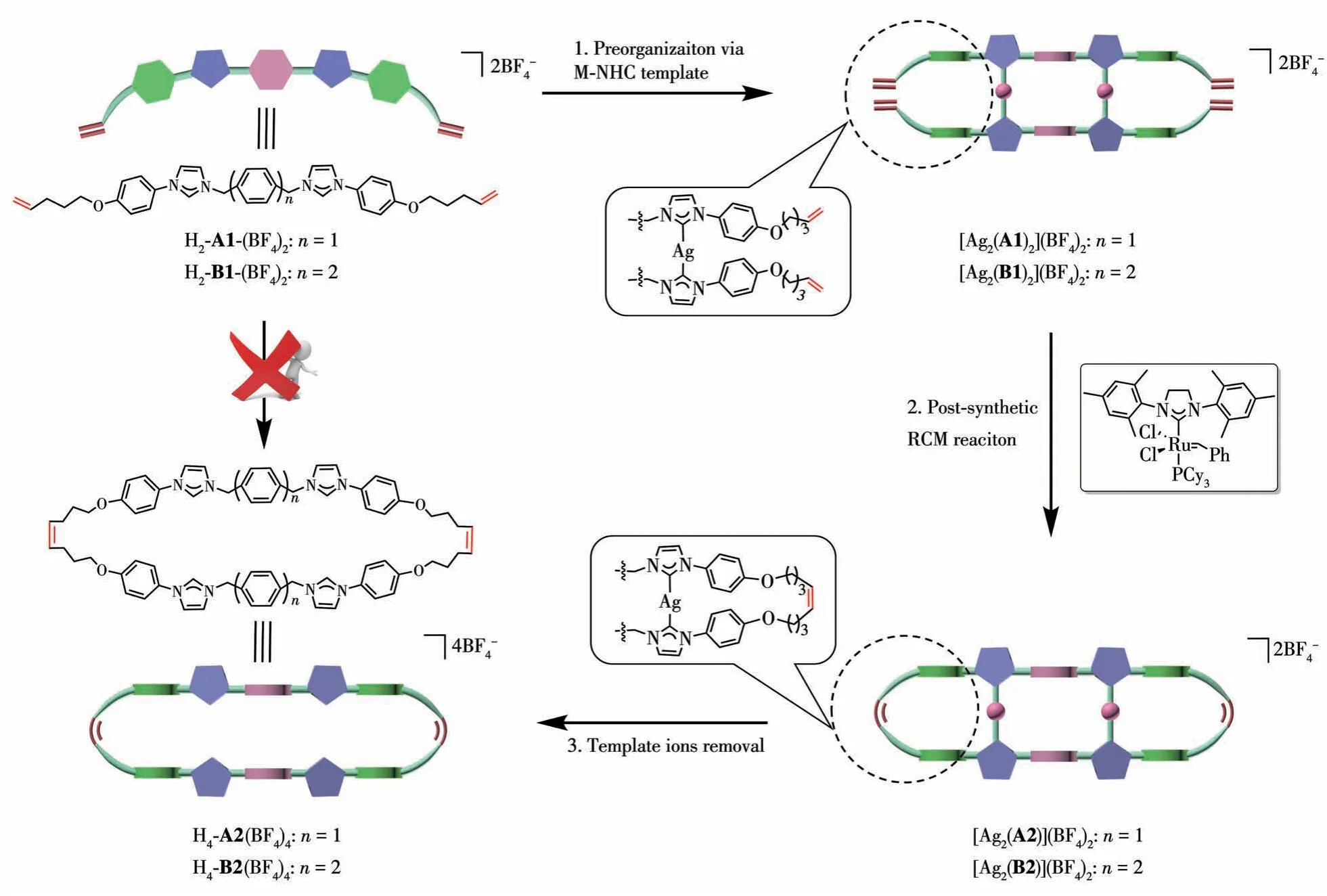
Scheme 1 Synthesis of the designed covalent organic macrocycles through metal-carbene-templated ring-closing metathesis approach
To facilitate this, we designed bidentate ligands H2-L1(BF4)2(L1=A1, B1), where two terminal olefins are connected to benzene rings via alkoxy linkers.For instance,H2-B1(BF4)2was synthesized using 4-(imidazol-1-yl)phenol, 4,4′-bis(bromomethyl)biphenyl, and 5-bromo-1-pentene,followed by exchanging the counteranion from bromide to tetrafluoroborate with NH4BF4.This anion exchange significantly improved the solubility of the ligand precursor in acetonitrile.The1H NMR spectrum of H2-B1(BF4)2in DMSO-d6(Fig.S1) reveals the characteristic resonance of the imidazolium NCHN proton atδ=10.01, consistent with previously reported examples[16-21].
The reaction of H2-L1(BF4)2(L1=A1, B1) with Ag2O yielded complexes [Ag2(L1)2](BF4)2(L1=A1, B1)in satisfactory yields.A thorough characterization was carried out utilizing various techniques, including NMR spectroscopy (1H,13C, H-H COSY, HSQC, and HMBC) and mass spectrometry (Fig.S4-S9).In the1H NMR spectra of [Ag2(B1)2](BF4)2, the expected resonance for the carbene ligand was observed, accompanied by the disappearance of the original imidazolium NCHN proton signal.Notably, characteristic doublet resonances for protons Hband Hcwere observed atδ=6.01-5.71 and 5.15-4.90, respectively (Fig.1a and 1b).In the13C and HMBC spectra, resonances corresponding to the CNHCatom and C=C carbon nuclei (Cb, Cc)were observed atδ=178.1, 137.8, and 115.3 (Fig.S5),respectively.Further validation through ESI-MS revealed two intense peaks consistent with the Ag(I)metallacycle, withm/zvalues of 1 571.445 1 and 742.255 2, corresponding to the [Ag2(B1)2(BF4)]+(Calcd.m/z=1 571.476 1) and [Ag2(B1)2]2+ions (Calcd.m/z=742.235 8),respectively(Fig.S9).
The pivotal step in our procedure was the templatedirected RCM, which proved highly suitable for creating the required rectangular structure.This reaction was conducted at a relatively low concentration of 0.6 mmol·L-1to prevent undesired side-oligomerization reactions.The metallacycle [Ag2(B1)2](BF4)2in anhydrous CH2Cl2was stirred at room temperature.Subsequently, a solution of Grubbs′ 2nd catalyst in dry CH2Cl2was slowly added into the solution.The resulting mixture was stirred at 40 ℃overnight, exclusively leading to the formation of the dinuclear cyclized product [Ag2(B2)](BF4)2.Evidence confirming the successful formation of the cyclized structure was provided by1H,13C,2D NMR spectroscopy,and HR-ESI spectrometry (Fig.S10-S15).Upon the RCM reaction, signals at 5.62-5.35 and 130.2,corresponding to the newly generated doublet resonances for He(Fig.1c)and Ce,respectively,were observed in1H,13C NMR spectroscopy(Fig.S10, S11).The ESI-MS spectrum of the final solution revealed signals originating from the most intense peaks atm/z=1 515.391 9 and 714.205 6, consistent with a silver (Ⅰ)metallacycle of the formula [Ag2(B2)](BF4)2.It suggested the presence of a cyclized intramolecular species and confirmed that the isotopic distribution of the dinuclear cyclized metallacycle closely matched the theoretical spectra(Fig.S15).
The two silver ions can be successfully removed from the complex [Ag2(B2)](BF4)2through the addition of NH4Cl.Subsequent anion exchange reactions with NH4BF4yielded the desired tetraimidazolium macrocycle in approximately 80% yield.Compound H4-B2(BF4)4was comprehensively characterized using NMR spectroscopy, and all protons and carbons were fully assigned through 2D NMR measurements(1H-1H COSY,HSQC, and HMBC, Fig.S16-S21).ESI-MS spectrometry (Fig.1e) confirmed the presence of peaks consistent with the target covalent organic macrocycle at 695.317 2(Calcd.for [H4- B2(BF4)2]2+695.318 6), 434.548 7(Calcd.for [H4- B2(BF4)]3+434.544 3), 304.161 1(Calcd.for[H4-B2]4+304.157 0).
To explore the ion detection capability of H4-B2(BF4)4,halide ions (F-,Cl-,Br-,and I-)were introduced(using their sodium salts) for ultraviolet, fluorescence,and1H NMR titrations (Fig.2).As illustrated in Fig.2b,the addition of 10.0 equivalents of F-, Cl-and Br-to the solutions of H4- B2(BF4)4, respectively, did not induce significant changes in the ultraviolet spectra.However, adding an equivalent amount of I-resulted in a blue shift from the original Soret band (258 nm) to a new band with maximum absorption at 241 nm.The absorption intensity increased significantly with increasing I-concentration (Fig.2c).Additionally, fluorescence titration experiments were conducted.No noticeable changes in the fluorescence spectra were observed upon adding 10.0 equivalents of F-, Cl-and Br-to the H4-B2(BF4)4solutions.However, upon the addition of I-,the fluorescence intensity of H4-B2(BF4)4at approximately 413 nm noticeably decreased (Fig.2d).As confirmed by1H NMR titration, the continuous addition of I-to the solution of H4-B2(BF4)4caused a downfield shift in the signal corresponding to the imidazolium Haproton (Fig.2e).No such significant changes were observed for the other protons, indicating that iodides bind to the cavity of the polyimidazolium macrocycle through hydrogen bonding involving C—H…I interactions(Fig.2a).
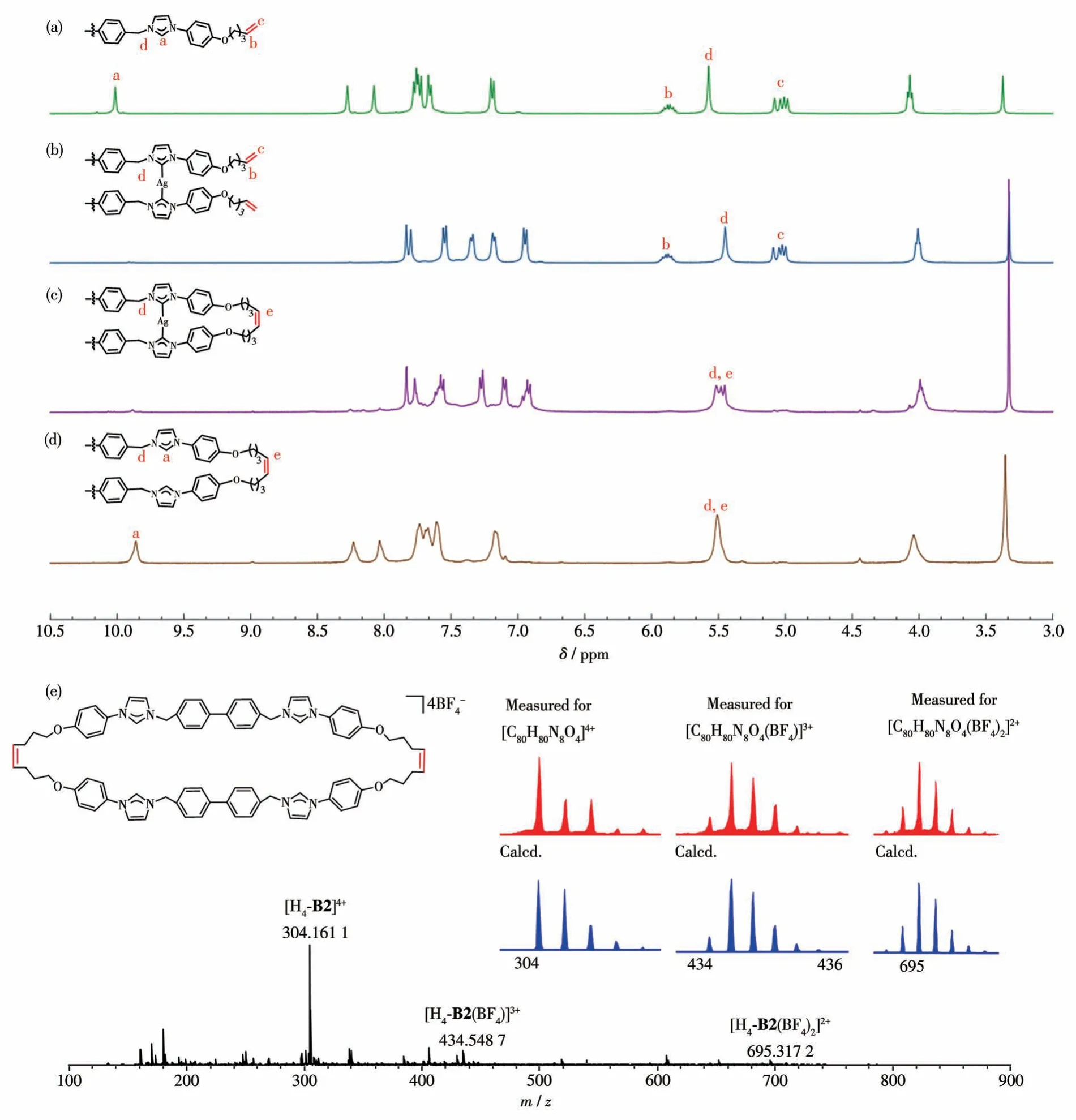
Fig.11H NMR spectra(400 MHz,DMSO-d6)of(a)H2-B1(BF4)2,(b)[Ag2(B1)2](BF4)2,(c)[Ag2(B2)](BF4)2,and(d)H4-B2(BF4)4,(e)HR-ESI mass spectra(positive ions)of H4-B2(BF4)4
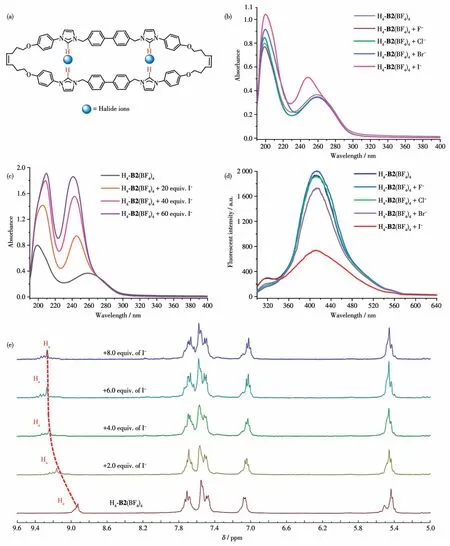
Fig.2 (a)Observed binding interactions between host H4-B2(BF4)4 and iodides,(b)Ultraviolet spectra of H4-B2(BF4)4(c=4×10-6 mol·L-1)and adding 10.0 equivalent of different ions(F-,Cl-,Br-and I-)in acetonitrile,(c)Ultraviolet titration spectra of H4-B2(BF4)4(c=4×10-6 mol·L-1)with I-in acetonitrile,(d)Fluorescent spectra of H4-B2(BF4)4(c=4×10-6 mol·L-1,λ=275 nm)and adding 100.0 equivalent of different ions(F-,Cl-,Br-and I-)in acetonitrile,(e)Partial 1HNMR spectra obtained upon continuous addition of NaI to a CD3CN solution containing H4-B2(BF4)4
3 Conclusions
In summary, we have demonstrated an efficient macrocyclization strategy for synthesizing covalent organic macrocycles through ring-closing metathesis employing a metal-carbene template.Our approach offers an alternative method for generating a silver biscarbene compound through the self-assembly of Ag(I)ions and dicationic bis-carbene precursors.This method effectively creates the required macrocyclic structure and facilitates the development of functionalized metallacycles.Utilizing the excellent properties ofNheterocyclic carbenes, the product can be easily isolated following the RCM reaction and Ag(I) removal,achieved through anion exchange reactions.Furthermore, this approach holds promise for preparing a wide range of functional organic macrocycles and threedimensional cages.Future endeavors will explore more intricate structures and properties in this context.
Conflicts of interest:All authors claimed no competing interest.
Acknowledgments:The current work was financially supported by the National Natural Science Fund of China(Grants No.22025107,22301240), Shaanxi Fundamental Science Research Project for Chemistry&Biology(Grants No.22JHZ003,22JHQ008), the Natural Science Basic Research Plan in Shaanxi Province (Grants No.S2023-JC-QN-1639,2022JQ-093),the National Youth Top-notch Talent Support Program of China,Xi′an Key Laboratory of Functional Supramolecular Structure and Materials,and the FM&EM International Joint Laboratory of Northwest University.
Supporting information is available at http://www.wjhxxb.cn
