肺缺血-再灌注核心基因介导的竞争性内源性RNA网络的构建
2024-01-19李晓凤唐明政刘禧禧宋子晴张国欣杨开银张凌云
李晓凤 唐明政 刘禧禧 宋子晴 张国欣 杨开银 张凌云
自1983年以来,肺移植已成为治疗各种终末期肺病的有效方法,全球每年约有4 000 例双肺或单肺移植[1-2]。肺移植目前存在的主要问题是供者短缺[3]。据报道,约24%等待供肺的患者可能死亡[4]。肺缺血-再灌注损伤(lung ischemia-reperfusion injury,LIRI)是导致肺移植失败和病死率高的主要原因[5],包括局部缺血性损伤和再灌注损伤[6]。现有改善LIRI 的治疗方案主要包括支持和替代治疗[7-9]。对寻找生物标志物、干细胞疗法、药物治疗和基因治疗等干预措施还在探索中[10],很少有用于临床的预防和治疗策略。因此,仍需要探索新策略预防LIRI,改善肺移植后的短期和长期预后。
竞争性内源性RNA(competitive endogenous RNA,ceRNA)是含有微小RNA(micro RNA,miRNA,miR)识别元件的RNA[11],可以通过与miRNA 竞争性结合来调节含有相应识别元件的基因或蛋白质表达[12]。其中环状RNA(circRNA)可通过反剪形成闭环,保护其免受核糖核酸酶R 的侵害,并稳定表达[13]。根据序列起源,circRNA 可分为3 类:外显子circRNA,内含子circRNA 和外显子-内含子circRNA[14]。长链非编码RNA(long non-coding RNA,lncRNA)可以“海绵”互补miRNA,促进其靶分子的去抑制,在lncRNA-miRNA-mRNA ceRNA网络中发挥作用[12,15]。ceRNA 网络可能会影响疾病并解释疾病过程,为新疗法提供思路[12]。因此,有必要在L I R I 中发现新的功能性以l n c R N A 介导的lncRNA-miRNA-mRNA ceRNA 网络,以确定更多与LIRI 诊断和预后相关的ceRNA 网络。本研究整合了多种生物信息学分析工具,以探索LIRI 的核心基因、通路、靶向药物和ceRNA 构建,为其治疗提供新的策略和思路。
1 材料与方法
1.1 微阵列数据信息
基因表达综合(Gene Expression Omnibus,GEO)数据库是一个提供高通量基因表达数据的公共基因组学数据存储库[16]。从GEO 数据库下载原始数据GSE145989 作为训练集,其中包括67 个人肺冷缺血样本和67 个人肺再灌注样本。将GSE172222 和GSE9634 数据集当作验证集,GSE172222 包括15 个人正常样本和16 个人LIRI 样本,GSE9634 包括6 个大鼠正常样本和12 个大鼠LIRI 样本。
1.2 DEG 的识别
使用LIMMA 包(http://www.bioconductor.org/packa ges/release/bioc/html/limma.html)分析微阵列数据,比较肺缺血与肺再灌注组织的表达值,以识别差异表达基因(differentially expressed gene,DEG)。在t检验后,将P值进行调整,并设定P<0.05 且对数折变率(logFC)>1 作为筛选标准。使用热图包(https://bioconductor.org/packages/release/bioc/html/he atmaps.html)绘制DEG 热图。
1.3 基因本体、通路富集分析
DAVID 基因功能分类工具可将大基因分类为生物模块[17]。基因本体(gene ontology,GO)是主要用于注释基因并分析其生物过程的生物信息学工具,而京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)则是一种数据库资源,可从高通量实验技术生成的大规模分子数据集中了解高级功能和生物系统。本研究使用clusterProfiler软件包(https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html)对DEG 进行GO 和KEGG 通路分析。GO 分析包括分子功能(molecular function,MF)、细胞成分(cellular component,CC)和生物学过程(biological process,BP),而通路分析则通过KEGG 对DEG 进行分类。本研究设置P<0.05 和基因计数>5 为临界点。
1.4 蛋白质-蛋白质相互作用网络集成
STRING 数据库是一个在线资源,其主要功能是构建功能性蛋白质关联网络[18]。本研究定义交互作用评分>0.9(中等可信度)为显著性。使用Cytoscape软件构建了蛋白质-蛋白质相互作用(protein-protein interaction,PPI)网络,并进一步分析DEG 之间的相互作用关系[19]。
1.5 筛选并验证核心基因
通过MCODE 插件选择候选核心基因,采用RStudio 中的受试者工作特征(receiver operating characteristic,ROC)软件包绘制ROC 曲线,对核心基因进行分析[20]。曲线下面积(area under the curve,AUC)>0.6 和P<0.05 作为核心基因的判定标准。
1.6 通过CIBERSORT 分析进行免疫浸润
采用CIBERSORT 算法分析之前获得的基因表达数据,以获取22 种免疫细胞的比例,其中包括幼稚B 细胞、记忆B 细胞、浆细胞、CD8+T 细胞、幼稚CD4+T 细胞、静息记忆CD4+T 细胞、活化记忆CD4+T 细胞、滤泡辅助性T 细胞、调节性T 细胞(regulatory T cell,Treg)、γδT 细胞、静息自然杀伤(natural killer,NK)细胞、活化NK 细胞、单核细胞、M0 型巨噬细胞、M1 型巨噬细胞、M2 型巨噬细胞、静息树突状细胞、活化树突状细胞、静息肥大细胞、活化肥大细胞、嗜酸性粒细胞和中性粒细胞。使用P<0.05 筛选样品,并计算各种免疫细胞在样品中的百分比。利用R 版本4.2.1 中的vioplot 软件包,分析免疫细胞的免疫浸润水平。
1.7 ceRNA 网络的构建和验证
通过starBase 检测和鉴定5 个核心基因预测的mRNA-miRNA 相互作用。从美国国家生物技术信息中心获取mRNA 序列,使用miRbase 下载人类miRNA 序列,使用miRanda 数据库预测mRNAmiRNA 的核酸结合。使用TargetScan 数据库预测miRNA 靶基因。StarBase 用于筛选mRNA-lncRNA相互作用,构建mRNA-miRNA-lncRNA 的ceRNA 网络。验证ceRNA 网络成员在GSE145989、GSE172222和GSE9634 中的表达。
1.8 基于ceRNA 网络的靶向药物
使用DGIdb 数据库分析可能靶向标记基因的药物,并基于ceRNA 网络成员,构建并分析它们之间的相互作用关系。
1.9 统计学方法
对所有的基因数据进行了对数转换以进行归一化处理,并使用R 软件(版本4.2.1)对数据进行可视化。P<0.05 为有统计学意义。
2 结果
2.1 DEG 的识别及GO、KEGG 和免疫细胞浸润分析
人LIRI 上调的DEG 基因101 个,下调的DEG 基因197 个。按调整后的P值排序的前100 个基因在各样本中的表达量通过热图显示(图1A)。
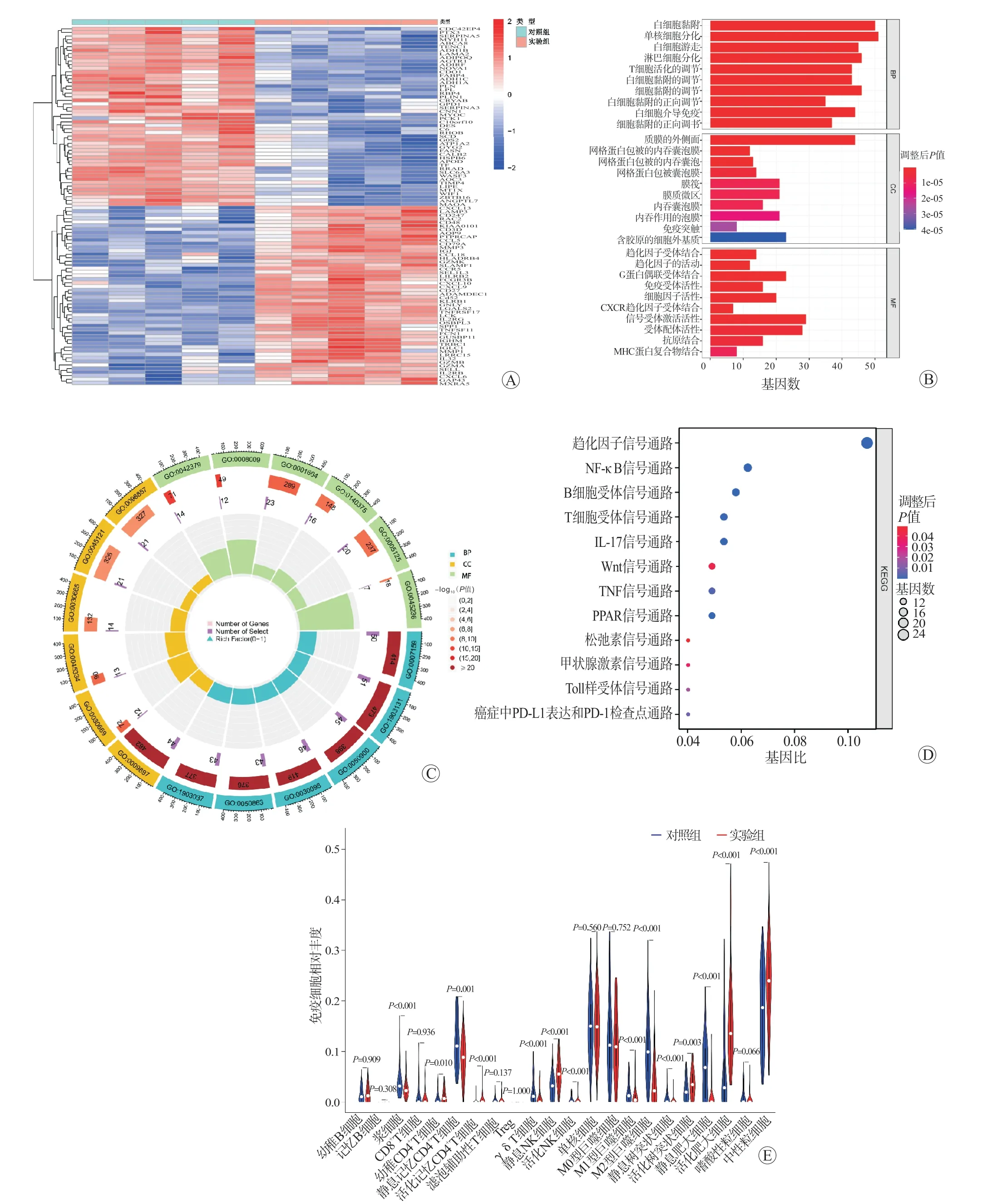
图1 DEG 的GO、KEGG 和免疫细胞浸润分析Figure 1 GO,KEGG and immune cell infiltration analysis of DEG
GO 分析结果显示,DEG 的BP 主要与细胞迁移、分化和调节有关;CC 主要与内吞囊泡膜、浆膜和膜筏有关;MF 主要与趋化因子受体、G 蛋白偶联受体、免疫受体活性和抗原结合有关(图1B、C)。
KEGG 分析结果显示,DEG 参与趋化因子、甲状腺激素、松弛素、聚腺苷二磷酸核糖聚合酶(poly adenosine-diphosphate-ribose polymerase,PPAR)、肿瘤坏死因子(tumor necrosis factor,TNF)、Wnt、白细胞介素(interleukin,IL)-17、T 细胞受体、B 细胞受体、核因子(nuclear factor,NF)-κB 等信号通路,及癌症中的程序性细胞死亡蛋白配体1(programmed cell death protein-ligand 1,PD-L1)表达和程序性细胞死亡蛋白1(programmed cell death protein 1,PD-1)检查点通路(图1D)。
免疫细胞浸润分析表明DEG 与浆细胞、幼稚T 细胞、静息记忆CD4+T 细胞、活化记忆CD4+T 细胞、γδT 细胞、静息NK 细胞、活化NK 细胞、M1 型巨噬细胞、M2 型巨噬细胞、静息树突状细胞、活化树突状细胞、静息肥大细胞、活化肥大细胞和中性粒细胞有关(图1E)。
2.2 核心基因的筛选
PPI 网络共获得了179 个节点,包括118 个上调基因和61 个下调基因组成的DEG(图2A),差异基因打分结果见图2B。CD8A、IL2RG、STAT1、CD3G 和SYK 为能够区分肺缺血和再灌注的候选核心基因,在高表达组中,这5 个基因呈上调状态(图2C)。5 个基因之间呈正相关关系,具有功能相似性(图2D)。
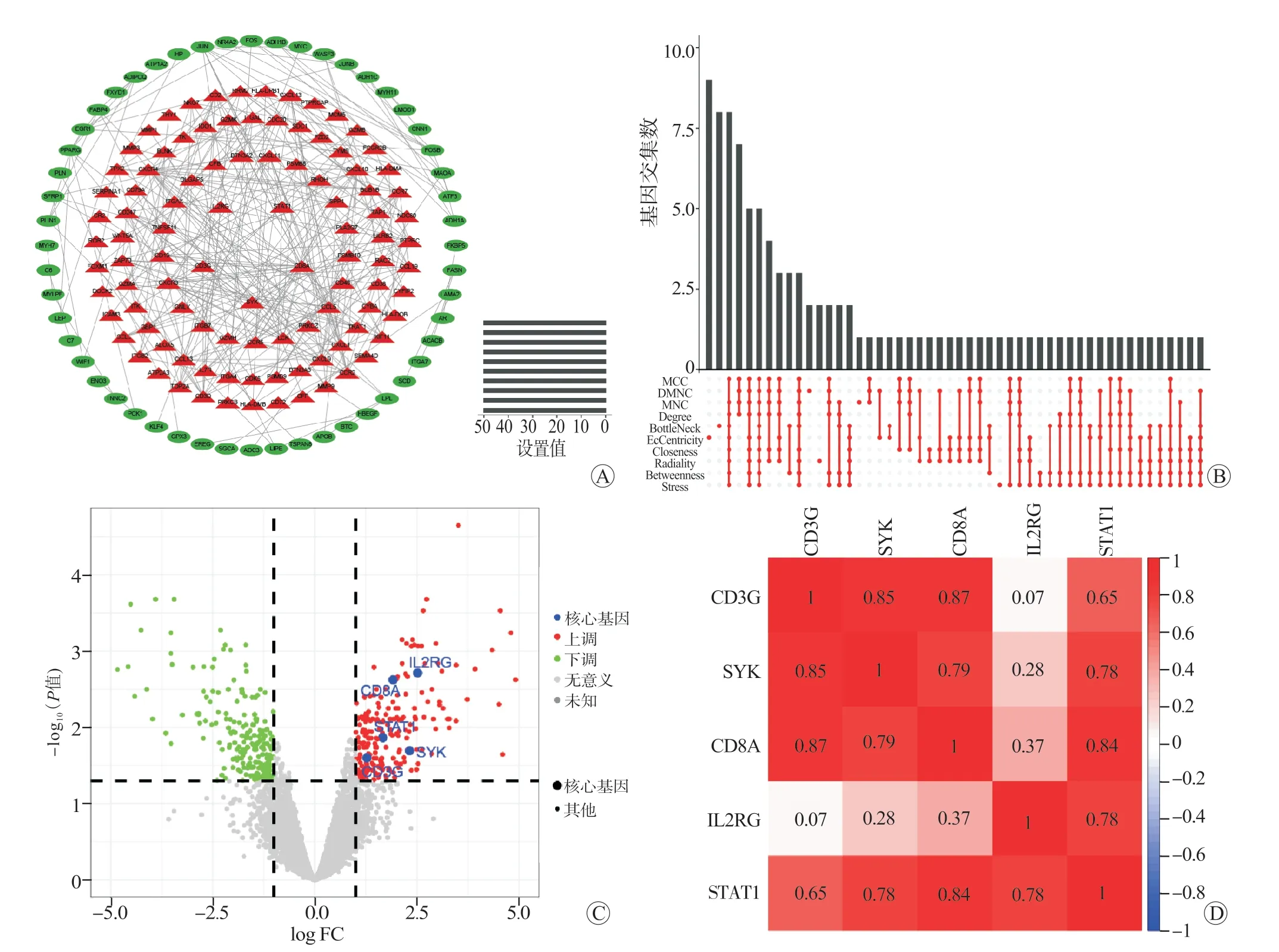
图2 核心基因的筛选Figure 2 Screening of the core genes
2.3 核心基因的诊断作用
CD8A、IL2RG、STAT1、CD3G 和SYK 5 个核心基因在训练集LIRI 组高表达(图3A),在验证组数据集里也高表达(图3B、C)。ROC 曲线结果显示这5 个核心基因诊断LIRI 具有相对较强的效力,但是CD8A 在鼠源性的LIRI 诊断效力比人源性的LIRI 更强(图3D、E)。

图3 核心基因的验证Figure 3 Validation of core genes
2.4 核心基因和免疫细胞相关性分析
SYK 与静息树突状细胞,M0 型巨噬细胞和滤泡辅助性T 细胞呈正相关,与活化树突状细胞和浆细胞呈负相关;STAT1 与M1 型巨噬细胞呈正相关;IL2GR 与中性粒细胞和活化记忆CD4+T 细胞呈正相关,与活化树突状细胞、M2 型巨噬细胞、静息肥大细胞和静息记忆CD4+T 细胞呈负相关;CD8A 与活化记忆CD4+T 细胞,CD8+T 细胞和滤泡辅助性T 细胞呈正相关,与幼稚C D 4+T 细胞呈负相关;CD3G 与M1 型巨噬细胞和CD8+T 细胞呈正相关,与记忆B 细胞、活化树突状细胞和幼稚CD4+T 细胞呈负相关(图4)。
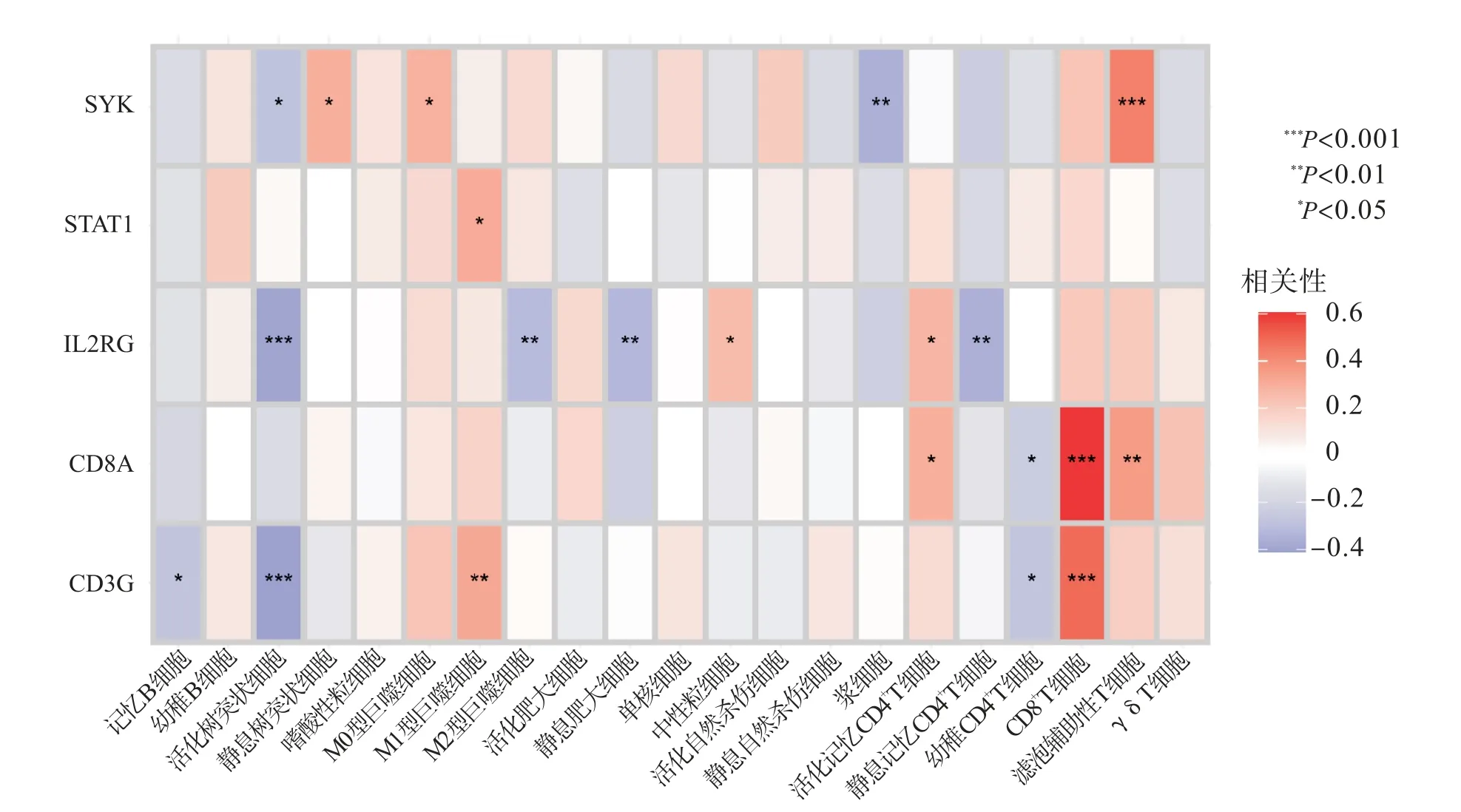
图4 核心基因和免疫细胞相关性分析Figure 4 Correlation analysis of core genes and immune cell
2.5 ceRNA 网络构建与表达验证
ceRNA 网络包括205 个节点(5 个核心基因,98 个miRNA,102 个lncRNA),217 个边缘(图5A)。共5 个lncRNA 可以竞争性地结合miR-146a-3p,10 个lncRNA 可以竞争性地结合miR-28-5p 和5 个lncRNA 可以竞争性地结合miR-593-3p 共同控制CD3G 的表达;19 个lncRNA 可以竞争性地结合miR-149-3p,8 个lncRNA 可以竞争性地结合miR-342-5p,6 个lncRNA 可以竞争性地结合miR-873-5p 和5 个lncRNA 可以竞争性地结合miR-491-5p 共同调节IL2RG;3 个lncRNA 可以竞争性地结合miR-194-3p 和miR-512-3p,5 个lncRNA 可以竞争性地结合miR-377-3p 和8 个lncRNA 可以竞争性地结合miR-590-3p 共同调节SYK;1 个lncRNA 可以竞争性地结合miR-590-3p 和4 个lncRNA 可以竞争性地结合miR-875-3p 共同调节CD8A;6 个lncRNA 可以竞争性地结合miR-143-5p,4 个lncRNA 可以竞争性地结合miR-1 231,8 个lncRNA 可以竞争性地结合miR-590-3p 和4 个lncRNA 可以竞争性地结合miR-875-3p 共同调节STAT1。
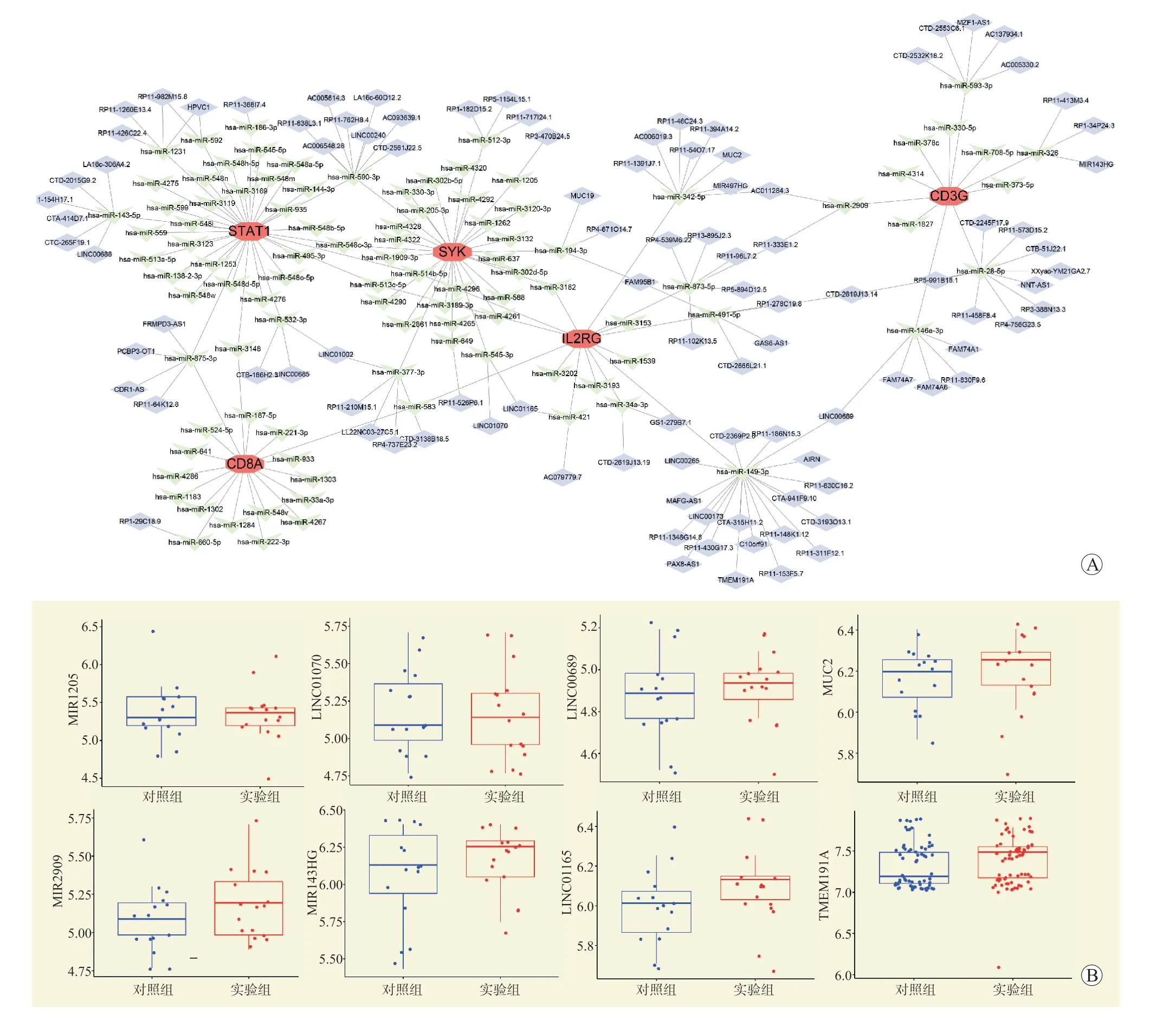
图5 ceRNA 网络构建与表达验证Figure 5 ceRNA network construction and expression validation
基于c e R N A 成员在数据集G S E 1 4 5 9 8 9、GSE172222 和GSE9634 的表达进行验证,结果发现MUC2、MIR2909、MIR143HG、LINC01165、MIR1205、LINC01070、LINC00689 和TMEM191A均为高表达(图5B)。
2.6 确定靶向药物
IL2RG 有4 种靶向药物,CD3G 有13 种靶向药物,SYK 有28 种靶向药物,lncRNA MUC2 有3 种靶向药物,未发现预测核心基因CD8A、STAT1 和其他ceRNA 网络基因的靶向药物。
赛度替尼、PRT-2 607、福坦替尼、R-343、ENTOSPLETINIB、R-112、R-406、HMPL-523、T A K-6 5 9、R-3 4 8 和R-3 3 3 是S Y K 的抑制剂(图6A);爱欧山和FORALUMAB 是CD3G 的抑制剂(图6 B);达利珠单抗和巴利昔单抗是IL2RG 的抑制剂,而阿地白介素是IL2RG 的受体激动剂(图6C);lncRNA MUC2 的靶向药物有3 种(图6D)。

图6 标记基因靶向药物的预测Figure 6 Prediction of marker gene-targeted drugs
3 讨论
肺移植是晚期肺病患者的有效治疗方法,但供肺的利用率较低[21]。LIRI 是肺移植术后早期原发性移植物功能障碍的最常见危险因素之一[22],并且会增加移植物功能障碍和排斥反应等风险[23]。尽管肺移植完全缺氧的时间只持续几个小时,却产生无法避免的影响[1]。严重的LIRI 可导致原发性移植物功能障碍,这是肺移植术后短期和长期病死率增加的主要原因[24]。目前尚无特定预防LIRI 的治疗方法,本研究结合了微阵列基因表达谱和生物信息学方法,旨在探索人类LIRI 过程中潜在新核心基因和分子机制。
首先,从GSE145989 数据集筛选DEG,并进行GEO、KEGG 和PPI 网络分析,筛选出61 个下调和118 个上调的DEG。通过Cytoscape 鉴定出5 个核心基因并进行ROC 曲线、免疫细胞相关性、ceRNA 网络构建等一系列分析。GO 分析显示DEG 与内皮细胞、G 蛋白偶联受体和免疫相关细胞等有关。有研究发现内皮祖细胞通过内皮型一氧化氮合酶途径减轻肺移植术后的缺血损伤[25],LIRI 后肺组织中S1P G 蛋白偶联受体1 上调表明S1P/S1PR1 轴参与LIRI 的病理生理过程[26],CXCR2 上调内皮细胞对肾缺血-再灌注损伤(ischemia-reperfusion injury,IRI)具有保护作用[27],而单核细胞、淋巴细胞、白细胞和T 细胞浸润易发生于LIRI 中[28]。KEGG 分析表明DEG 参与包括NF-κB、PPAR、TNF、Wnt、B 细胞受体和Toll 样受体等信号通路,各通路在LIRI 都扮演着重要的角色。如甲烷和西洛他唑可分别通过磷脂酰肌醇-3-激酶(phosphatidylinositol 3-kinase,PI3K)-蛋白激酶B(protein kinase B,Akt)-NF-κB 和PPARA 信号通路调节缺氧-复氧引起的氧化应激、炎症因子释放和细胞凋亡,促进LIRI 修复[29-30]。此外,miR-145 可以NF-κB 依赖的方式抑制Beclin1 和SIRT1,从而减弱LIRI 小鼠模型自噬过程[31]。B 细胞依赖性途径则在小鼠IRI 后慢性肺同种异体移植排斥反应中发挥作用[32]。TNF-α 激活JNK/FoxO3a 轴,延迟中性粒细胞凋亡并导致LIRI 发展[33]。另外,miR-122 通过Toll 样受体信号通路加速LIRI[34]。
本研究发现,与LIRI 相关的5 个核心基因分别为CD8A、IL2RG、STAT1、CD3G 和SYK,ROC 分析验证其具有较高的诊断价值。值得注意的是,已有研究证实IL2RG、SYK 和CD8A 与IRI 相关。如IL2RG 小鼠通过减少中性粒细胞损耗来降低肝脏IRI[35]。而抑制SYK 线粒体裂解途径与IRI 微血管保护有关[36]。另一项研究显示,CD8A 在肾IRI 过程中表达最高[37]。此外,本研究还发现巨噬细胞与SYK、STAT1 和CD3G 呈正相关,T 细胞及其相关细胞与SYK、IL2GR、CD8A 和CD3G 呈正相关,SYK 与树突状细胞和IL2GR 与中性粒细胞呈正相关。有研究发现,巨噬细胞和树突状细胞中的Mincle-Syk 通路与微生物群调节能够影响IL-17 和IL-22 表达[38-39]。但在B 细胞和T 细胞中,SYK 的表达在整个进化过程中是严格分离的[40]。此外,STAT1 促进巨噬细胞内Nampt 表达和功能,并且增强巨噬细胞的抗病毒先天免疫[41-42]。CD8A 通常主要在效应性T 细胞和细胞毒性T 淋巴细胞上表达,但有时也在Treg、NK 细胞以及树突状细胞上表达[43-45]。
ceRNA 网络分析揭示了核心基因与众多的miRNA 和lncRNA 复杂关系,包括miR-146a-3p、miR-149-3p、miR-28-5p、miR-342-5p、miR-377-3p、miR-590-3p、MUC2 和miR143HG 等miRNA 和lncRNA。基于ceRNA 网络分析结果发现miRNA 已有研究。miR-146a-3p 抑制NF-κB 通路保护心脏和小肠免受IRI[46-47],并在急性肺损伤中减轻炎症反应[48]。miR-149-3p 与心肌IRI 有关[49],而miR-28-5p 与脊髓IRI 也有关[50]。神奇的是,肺源性外泌体中的miR-28-5p 在急性肺损伤中也调节间充质干细胞的功能[51]。此外,lncRNA PEG11as 通过miR-342-5p/PFN1通路加重脑IRI[52],而miR-342-5p 调控GPRC5A 通路预防心肌IRI[53]。另外,miR-342-5p 和miR-590-3p 分别抑制TLR9 和TRAF6 以减轻败血症小鼠的急性肾损伤[54-55],而miR-377-3p 靶向RPTOR 诱导自噬,从而改善脂多糖诱导的急性肺损伤[56]。下调mmu_circ_0000943 则通过调节mmu-miR-377-3p/Egr2 轴,改善肾IRI 引发的炎症和氧化应激[57]。此外,miR-590-3p 通过调节HMGB1/TLR4/MyD88/NF-κB 轴来防止氧-葡萄糖剥夺和复氧细胞模型中的IRI[58]。最后,MUC2 黏蛋白和非黏蛋白微生物群在肠损伤中具有特殊的宿主防御作用[59],而miR143HG通过miR-504 海绵效应和miR-21 甲基化来抑制细胞的增殖[60]。
综上所述,本研究通过挖掘微阵列基因表达谱和生物信息学相结合,发现了5 个核心基因,为研究LIRI 的分子机制和治疗靶点提供了新的思路和切入点。本研究运用基因表达谱和生物信息学相结合的方法发现人类LIRI 过程中的潜在新核心基因和ceRNA,但由于基因芯片数据的可用性有限及满足研究条件的样本数较少,本研究纳入的样本量有限,因此仍需要在更多的实验和临床实践中进一步验证研究结果。
