流动注射-亚甲基蓝分光光度法测定水中阴离子表面活性剂的方法改进研究
2024-01-16王筱径
王筱径
(福建省地质测试研究中心,福州,350001)
近年来,伴随着经济发展和人民生活质量的提高,阴离子表面活性剂被广泛地运用于生产生活的各个方面,为人们带来了许多便利。然而,大量阴离子表面活性剂随生活污水或工业废水流入水体,会导致水质恶化,对生态环境造成破坏。研究表明,当水体中阴离子表面活性剂浓度达到0.5 mg/L时,会阻断水体中的氧气交换,造成微生物大量缺氧死亡[1]。阴离子表面活性剂还具有难降解性,能与水中的其他污染物结合在一起形成分散性的胶体颗粒,增大污染的浓度和毒性[2]。因此,阴离子表面活性剂成为反映水环境质量的重要指标之一,是国家采测分离“9+x”中要求抽测的项目[3]。
目前,可用于测定水中阴离子表面活性剂的标准检测方法主要有“亚甲蓝分光光度法”[4]、“二氮杂菲萃取分光光度法”[5]、“流动注射-亚甲基蓝分光光度法”[6]、“连续流动分析-分光光度法”[7]等。其中,前二者方法具有灵敏度高、稳定性好、抗干扰能力强的优点,但操作步骤繁琐,系统误差大,且该类方法氯仿使用量大,容易对操作人员身体健康造成危害。而后二者方法为近年发展起来的仪器分析方法,与传统方法相比,分析效率高、测量准确、重复性好,对操作人员健康的危害较小。但使用标准方法“流动注射-亚甲基蓝分光光度法”进行样品测定时,仍然存在步骤繁琐、试剂消耗量大等问题,为了更好地开展样品检测工作,研究在标准方法的基础上进行一些改进,优化试剂的用量,简化实验溶液的配制方法。通过对改进的方法的标准曲线、检出限、精密度、正确度、加标回收率等特性指标进行确认,评估该方法在技术上的可行性、合理性。
1 实验方法
1.1 方法原理
将含有阴离子表面活性剂的试样注射到一个运行连续载流的管路中,让试样与实验溶液按照事先设定的顺序混合并发生化学反应,生成的亚甲基蓝活性物质被氯仿萃取并进入流通池进行检测分析。仪器根据亚甲基蓝活性物质浓度与吸光度的线性关系建立标准曲线,依据线性回归方程得出样品中阴离子表面活性剂的浓度。样品中存在的干扰物质在仪器中经过萃取和反萃取可被消除[8]。
1.2 方法改进
流动注射-亚甲基蓝分光光度法采用流动注射分析仪作为主要仪器,由于仪器结构精密、管路复杂,对实验溶液的使用要求较高,因此溶液配制过程步骤繁琐,试剂消耗量大。研究通过对标准方法进行改进,实现了试剂用量的优化和溶液配制方法的简化,使得流动注射分析法快速、准确、环保的技术优点能够得到充分体现。使用标准方法与使用改进的方法配制实验溶液的对比(表1)。
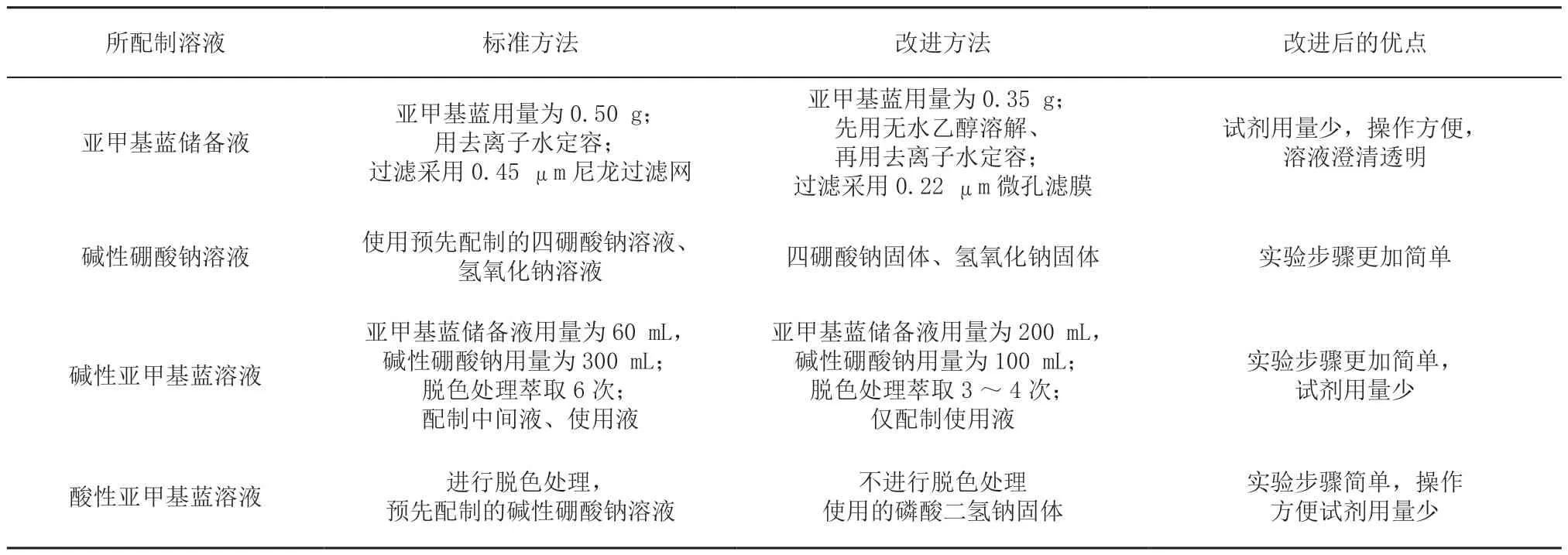
表1 标准方法与改进的方法配制实验溶液的对比Table1 Comparison of the standard methods and the modified methods for preparing the experimental solution
(1)亚甲基蓝储备液标准方法是将适量的亚甲基蓝溶解于1 L 去离子水中,用0.45 μm 尼龙过滤网过滤的方式制备亚甲基蓝储备液(亚甲基蓝浓度为0.50 g/L)。改进方法采用将亚甲基蓝溶解于500 mL 无水乙醇,再用去离子水定容,然后使用0.22 μm 微孔滤膜过滤的方法制备亚甲基蓝储备液(亚甲基蓝浓度为0.35 g/L)。改进的方法操作方便,且减少了亚甲基蓝的使用量,而采用孔径更细的过滤方式,过滤后得到的溶液澄清透明,在此后配制酸性、碱性亚甲基蓝溶液时使用效果更好。
(2)配制碱性硼酸钠溶液:标准方法采用分别配制四硼酸钠溶液和氢氧化钠溶液,再将2 种溶液等体积混合的方式。改进方法则采用四硼酸钠固体和氢氧化钠固体混合溶解进行配制。
(3)制备碱性亚甲基蓝溶液:标准方法需要配制2 种碱性亚甲基蓝溶液,并反复萃取6 次进行脱色,操作步骤繁琐,试剂消耗量大。改进方法简化了操作步骤,仅配制1 种碱性亚甲基蓝溶液,并萃取3 ~4 次进行脱色。同时增大了亚甲基蓝储备液的用量,由60 mL 增加到200 mL,确保样品中的阴离子表面活性剂能够全部参与反应并被萃取分离。
(4)制备亚甲基蓝储备液:标准方法酸性亚甲基蓝溶液需萃取3 次进行脱色,由于在一次萃取时样品中的阴离子表面活性剂已全被萃取分离,在二次萃取时,酸性亚甲基蓝溶液参与反应的作用只是反洗样品中的无机阴离子[9],消除干扰,所以改进方法配制酸性亚甲基蓝溶液时不进行脱色处理,节省了时间和成本。改进方法采用磷酸二氢钠固体代替碱性硼酸钠溶液作缓冲剂,操作起来更加方便。
2 实验材料与参数测定
试验的主要试剂有亚甲基蓝、四硼酸钠、三氯甲烷、硫酸、氢氧化钠、无水乙醇、磷酸二氢钠、石油醚,阴离子表面活性剂(十二烷基苯磺酸钠)标准溶液,浓度值为1 000 mg/L,标准物质编号为GBW(E)081623,批次编号为L5X2;实验用水为实验室制备的去离子水。
此次实验的仪器设备为BDFIA-8000 型全自动流动注射分析仪,北京宝德仪器有限公司生产。仪器的参数设置中的样品周期为200 s,泵转速为35 r/min,注射时间为78 s,进载流时间为78 s,到达阀时间为82 s。
设定仪器参数先用去离子水分别清洗载流,碱性亚甲基蓝、酸性亚甲基蓝的流路约为15 min。待清洗步骤结束后,将各个进试剂的毛细管插入相应的试剂瓶中,继续运行15 ~20 min,期间检查整个流路的密闭性及液体流动的顺畅性,待基线稳定后,编写进样列表,之后可以开始样品的测定。然后量取标准系列溶液、样品待测液置于流动注射分析仪进样器内,按顺序进行测定。
3 结果与分析
3.1 标准曲线的绘制
此试验配制阴离子表面活性剂浓度分别为0,0.10,0.20,0.50,1.00,2.00 mg/L。经自动进样测定,得到所配制标准系列的信号峰面积,标准曲线测试结果(表2)。以信号峰面积为横坐标,对应的标准溶液浓度为纵坐标绘制标准曲线,得到线性回归方程为y=0.001 738x+0.002 287,线性相关系数大于0.999,线性关系满足“流动注射-亚甲基蓝分光光度法”的质量控制要求。

表2 标准曲线测试结果Table 2 Standard curve test results
3.2 标准曲线的中间浓度点校准核查
选取浓度为0.50 mg/L 的标准曲线中间浓度点,每隔10 个样品进行校准测定,计算出校准点与标准曲线该点浓度的相对偏差。标准曲线的中间浓度点校准核查结果(表3)。
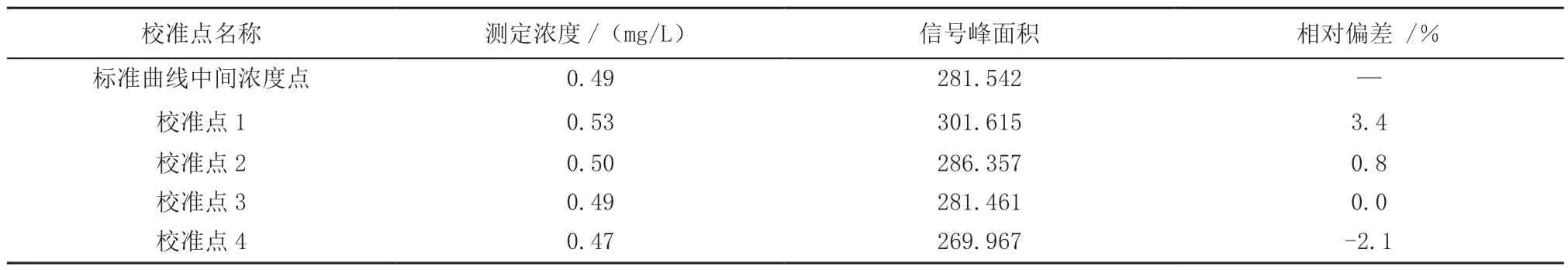
表3 标准曲线的中间浓度点校准核查结果Table 3 Results of the intermediate concentration point calibration verification of the standard curve
3.3 检出限的计算
对配制浓度约为估计方法检出限5 倍的标准溶液进行7 次平行测定,依据公式(1)计算出方法检出限MDL。
式中:n为样品的平行测定次数,为6;t为自由度,为n-1;置信度为99%的t分布值(单侧),本次取3.143;S为n次平行测定的标准偏差。
根据测试结果计算出MDL为0.03 mg/L,测定下限为4 倍的MDL,所得结果为0.09 mg/L,优于标准方法对检出限及测定下限的要求标准曲线。测试结果(表4)。
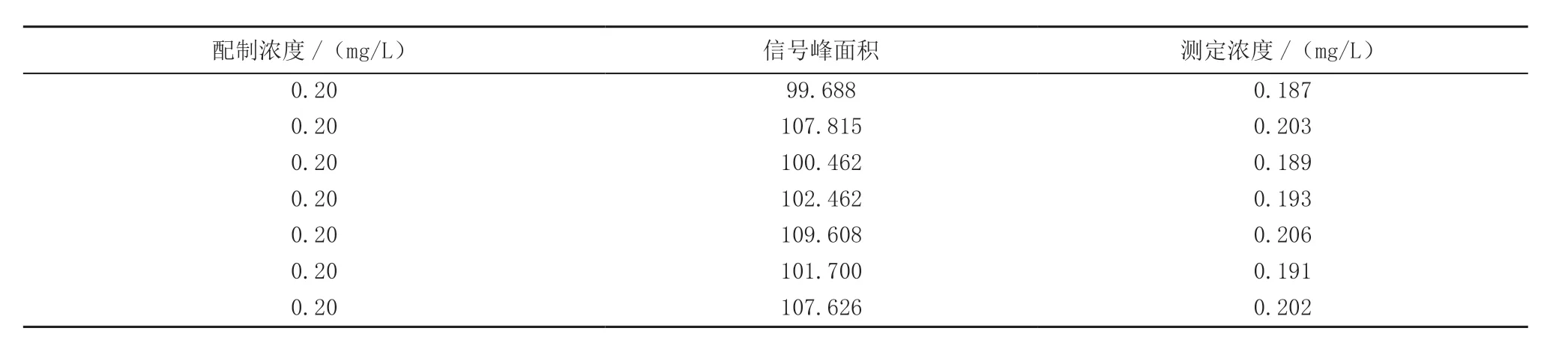
表4 方法检出限、测定下限的测试结果Table 4 Test results of the method detection limit and lower measurement limit
3.4 精密度实验
配制低、中、高3 种浓度的标准溶液,低浓度为0.40 mg/L,中浓度为1.00 mg/L,高浓度为1.80 mg/L。每种溶液连续测试6 次,精密度测试结果(表5)。根据测试结果计算出3 种样品的相对标准偏差分别为1.90%,0.89%,0.70%,数据不大于标准方法中6 家实验室所测得2.2%~6.9%、1.2%~6.1%、0.6%~6.7%的相对标准偏差范围,证明方法精密度良好。

表5 精密度的测试结果Table 5 Test results of the precision单位:mg/L
3.5 正确度实验
采用改进方法和标准方法分别进行有证标准物质的测定以及加标回收实验,通过分析实验结果评价方法的正确度。有证标准物质浓度为(0.391±0.029)mg/L,由生态环境部环境发展中心环境标准样品研究所生产,测试分析时取样品10 mL,实验室用水稀释至250 mL 后用于测试。标准方法测试结果为0.402 mg/L,改进方法的测试结果为0.399 mg/L,2 种方法的测试结果均在真值的浓度范围内。加标回收实验采用对3 种不同类型的实际样品加入同一加标浓度的方式,依据“环境监测分析方法标准技术导则”[10]中对加标浓度的规定(样品有检出时,加标浓度应为样品浓度的0.5 ~3 倍;样品未检出时,加标浓度应尽可能包含适用的生态环境质量标准、生态环境风险管控标准、污染物排放标准限制的浓度),确定加标量为0.40 mg/L。加标回收实验的测试结果(表6)。经改进方法测定,地表水和地下水样品的本底浓度为0,生活污水样品的本底浓度为0.21 mg/L,经样品加标回收实验可知,样品的加标回收率为87.5%~105.0%,该回收率与采用标准方法测定的回收率偏倚一致。由以上的实验结果可见,改进方法具有良好的正确度。

表6 实际样品加标测试结果Table 6 Test results of the actual sample-spiked
4 结论
(1)研究对“水质阴离子表面活性剂的测定”标准检测方法进行改进,优化了试剂的使用量,简化了实验溶液的配制方法。为评价改进后方法的特性指标是否满足标准方法的要求,对其进行了确认,测定了标准曲线、检出限、精密度、正确度、加标回收率等特性指标。由实验结果可知,标准曲线线性关系良好,线性相关系数大于0.999,满足方法规定的要求;校核点与标准曲线该浓度点的相对偏差为-2.1%~3.4%、满足要求的相对偏差应≤±10%。方法检出限为0.03 mg/L,测定下限为0.09 mg/L,优于标准方法对检出限及测定下限的要求,实验得出的精密度、正确度、加标回收率均满足标准方法的要求。
(2)采用改进方法和标准方法分别进行有证标准物质的测定以及加标回收实验,2 种方法的测试结果无明显差异。实验结果表明,改进后方法的特性指标能够满足标准方法的要求,技术上合理可行,且该方法最大限度地发挥了流动注射分析法快速、准确、环保的技术优点,具有一定的推广应用价值。
