1 例2p11.1-q21.1 片段重复导致智力障碍患儿的遗传学及表型分析
2024-01-10刘楠王萍徐晓薇王学韬武晋英李佳慈舒剑波
刘楠,王萍,徐晓薇,王学韬,武晋英,李佳慈,舒剑波
(天津市儿童医院(天津大学儿童医院);天津市儿科研究所;天津市儿童出生缺陷防治重点实验室,天津300134)
智力障碍以往被称为智力落后(MR),是一种不健全的智力发育,导致患者与相同年龄、性别和社会文化背景的个体相比,在一般心智能力、智力功能、适应性行为和功能技能方面存在明显缺陷。遗传学病因是导致智力障碍的重要因素,目前已发现1 700 多种基因与智力障碍相关[1]。智力障碍可以是多种遗传综合征的共同特征,也可能是孤立的发现,其相关的表型和遗传异质性明显。因而采用包括染色体核型分析、WES 等多种细胞及分子遗传学检测方法相结合,可以在早期识别智力障碍患儿的发病原因,帮助临床医生确认或建立临床诊断,为临床诊疗方案的选择及遗传咨询提供客观依据[2-3]。本文应用G 显带染色体核型分析和WES 诊断1 例由于2p11.1-q21.1 片段重复导致临床表现以智力障碍为主、伴发多系统发育畸形的患儿,现报告如下。
1 病例资料
1.1 一般情况 患儿,女性,14 周岁,因学习成绩落后于2019 年7 月11 日就诊于天津市儿童医院门诊。第1 胎第1 产,母亲孕期体健,足月剖宫产,否认生后窒息史。患儿14 个月可独立行走,幼儿期及学龄前期无特殊异常表现,自学龄期开始出现学习成绩落后,课上小动作多,社会交往能力欠佳,喜欢模仿他人言行,情绪易激动。11 岁月经初潮,周期规律。家族史无异常。
1.2 查体 身高146 cm,低于平均值2 SD,头围51 cm,提示生长迟缓、小头畸形;发际低,颈蹼,短耳畸形,双手食指尺侧偏斜(图1)。心、肺、腹部查体未见异常,第二性征已开始发育。
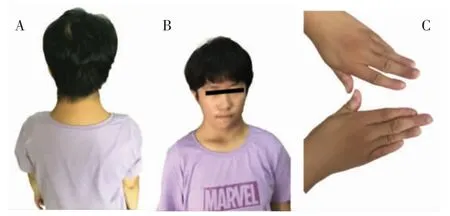
图1 患儿头面部及双手照片
1.3 特殊检查 骨龄X 线:左手掌指骨骨骺与干骺端均已闭合,左腕关节见8 枚腕骨,左侧拇指内收肌籽骨出现,左侧肘关节骨骺出齐并闭合,提示骨龄提前。韦氏儿童智力量表全式(C-WISC):IQ=61,诊断智力障碍;感觉统合能力:轻度失调;儿科患者心理社会风险评估(psychosocial risk assessment in pediatrics,PRAP)量表:提示中度风险(11 分);异常行为量表(aberrant behavior checklist,ABC):17分;视听整合连续测试(IVA-CPT):综合注意力商数=40。
1.4 染色体G 显带核型分析 经患儿家属知情同意,获取患儿及其父母外周血标本。本研究经本院伦理委员会批准(2016021)。应用细胞遗传学分析常规方法,对患儿血样进行淋巴细胞培养及中期染色体标本制备并进行G 显带染色制片。于显微镜下分析至少10 个中期分裂相,计数30 个分裂相,按照人类细胞遗传学国际命名体制(ISCN2016)标准进行染色体核型分析。结果显示患儿存在一个衍生的2 号染色体,核型为46,XX,der(2)(pter→q11.1::?::q12→qter),即2 号染色体长臂出现形态异常,但无法确定该片段的来源,患儿的父母核型均正常(图2),提示其衍生的2 号染色体为新发突变。
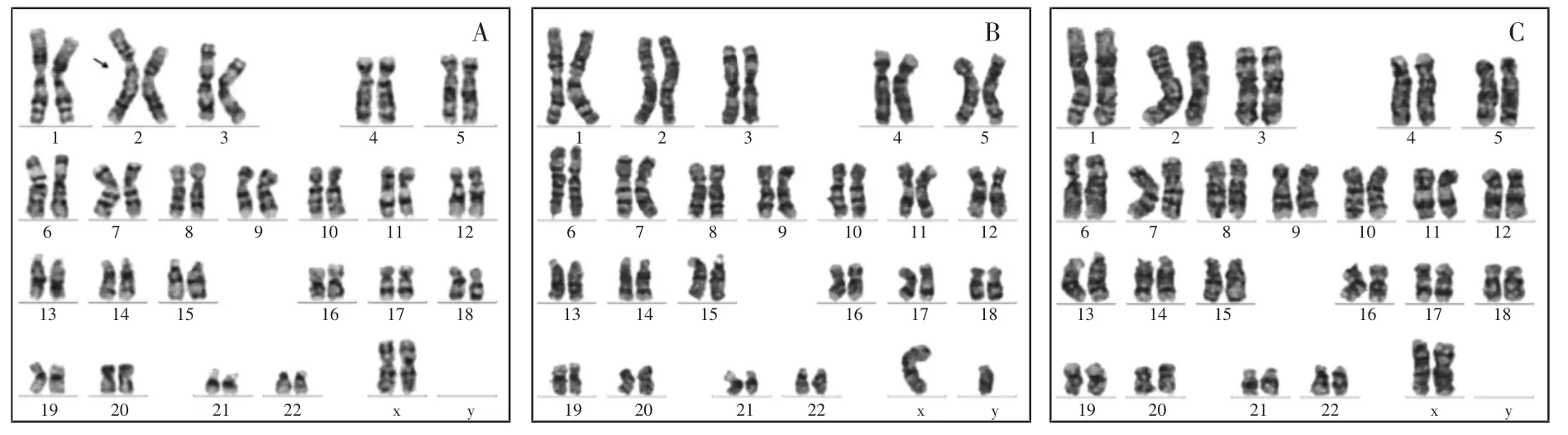
图2 染色体核型分析结果
1.5 拷贝数变异(CNVs)分析 采集患儿外周静脉血2 mL,加入内置有EDTA 抗凝剂抗凝的试管,提取基因组DNA 并构建文库。检测合格的DNA 文库利用Illumina 公司的Hiseq2500 二代测序仪进行测序,平均深度达到100~200 X。通过测序深度的对数转换推算CNVs,参考以下国际数据库:DECIPHER、OMIM、ISCA、UCSC、DGV、Clingen Dosage Sensitivity Map,利用生物信息学分析对WES 结果进行CNVs分析。结果为wes(hg19)2p11.1q21.1(91634679~131221727)×3(图3),回顾分析2 号衍生染色体片段重复带型特点,推断2p11.1p11.0 出现倒位插入重复,2q11.1q21.1 出现正向重复,因此患儿的最终核型被确定为46,XX,dup(2)(pter→cen→p11.1::q11.1→q21.1::q11.1→qter)。
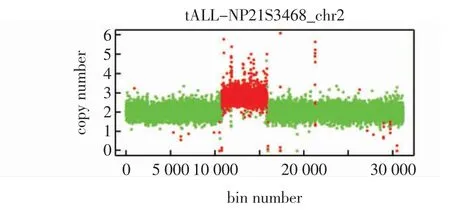
图3 患儿基于WES 结果的CNVs 分析结果图
2 讨论
基于不同人种的群体研究均表明,智力障碍的发生与遗传因素密切相关[4-5],因此,笔者首先对本例患儿进行外周血的染色体核型分析。经过对中期染色体形态的观察并计数,发现患儿其中1 条2 号染色体长臂上存在异常重复片段,为单一核型46,XX,der(2)。但由于G 显带核型图中每一条染色体是根据其大小以及深浅带型特征区分的,且患儿父母染色体核型均为正常,因而对于孤立出现的2 号染色体上的异常片段无法确定其来源和基因组特征[6]。而应用全外显子测序进行CNVs 分析的分子遗传学检测方法,弥补了细胞遗传学核型分析的技术局限性,可提供异常片段的基因内涵背景数据,为依赖于基因功能及其缺失或重复变异致病性的临床表型分析提供了客观依据[7-9]。据此,笔者通过患儿WES 数据结果基础上的CNVs 分析,确定重复片段的来源为2 号染色体p11.1-q21.1(91634679~131221727)。该片段大小约为39.59 Mb,区域内包含196 个编码基因,其中与疾病有关联的基因共41个。参照2020 年美国医学遗传学与基因组学学会(ACMG)联合临床基因组资源中心(ClinGen)发布的染色体CNVs 致病性评估标准,该变异符合3C(0.9 分,该区域包含大于50 个编码基因)与4M(0.3分,与对照相比,在病例中的频率显著增加,表型不够一致、特异),评分结果为1.2 分,判定为致病性变异[10]。
目前,对于2 号染色体p11.1-q21.1(91634679~131221727)片段内部分区域重复的病例多有报道。Wolfe 等[11]发现,2q13 片段重复患者的主要临床表现为智力障碍(70%)、注意力缺陷多动障碍(attention deficit hyperactivity disorder,ADHD,60%)、小头畸形(29%)、孤独症谱系障碍(autism spectrum disorder,ASD,17%)。有学者在2 419 例发育障碍患者中观察到,携带2q13 重复病例的CNVs 片段大小为1.4 ~2.1 Mb,且都有一个共同的1.3 Mb 区域(chr2:111449141~112746937,GRCh37,hg19),该变异在关于智力障碍、发育迟缓和神经精神疾病的研究中多有报道[12]。而Dharmadhikari 等[13]在临床表型为发育迟缓、ASD、癫痫及ADHD 的5 个家系中,发现了2q21.1 区域内的片段重复。这些拷贝数重复片段皆在本文病例异常片段区域内,只是本例患儿表现为智力发育迟缓、行为异常但没有癫痫的表现。这种相同的CNVs 却显示出不同临床表型的现象,与表观遗传学外显率密切相关。外显率是指一定环境条件下,群体中某一基因型个体表现出相应表型的百分率。有学者对5 026 例产前诊断结果进行统计分析后发现,人类常见的染色体重复/缺失的外显率,仅少部分为100%[14]。因而,考虑患儿没有出现癫痫表型可能与此密切相关。
尽管大多数CNVs 对患者临床表型有明显影响,但其致病机制仍不清楚。许多大型CNVs 包含数百万个核苷酸和几十个基因,混淆了它们的关键致病因素。甚至,具有相同CNVs 的患者可能出现“镜像”表型,例如对2q13 片段重复患儿[11]的临床表型统计发现,29%出现“小头畸形”,14%表现为“巨头畸形”,这些“镜像”表型是由CNVs[15-16]中包含的一个或多个、具有不同剂量敏感(dosage sensitive,DS)性的基因或元素所导致的。美国国立卫生研究院临床基因组资源中心(ClinGen)开发了标准框架体系,用以评估基因和疾病之间的关联性以及缺失(单倍剂量敏感)或重复(3 倍剂量敏感)的个体基因或基因组区域致病性证据的可信性强度[17]。本例患儿重复片段来源为2 号染色体p11.1-q21.1(91634679~131221727),该区域与疾病有关联的基因为41 个,各基因与疾病的剂量敏感特性不同,导致患儿具有以智力障碍为主伴有生长迟缓、神经精神缺陷、多发畸形的复杂临床表型。其中,AFF3基因为ATF 转录因子家族基因成员,是超级延伸复合体的组成部分。它调节神经发生和发育相关基因的表达,是Wnt/β-连环蛋白通路的靶点之一,也是参与骨发育和稳态通路的重要基因。OMIM 网站显示该基因异常可导致KINSSHIP 综合征,临床表现中包括智力障碍、生长发育迟缓、小头畸形、缩颌及掌指关节异常均与本患儿表型相符合,呈常染色体显性遗传模式。经ClinGen 网站检索,目前无足够证据证明其为三倍剂量敏感,但是已总结了11 例该基因缺失或重复病例,其中9 例确定为AFF3重复致病,两例缺失致病,其中显示3 倍剂量敏感病例的临床表型与本文患儿基本相符[18-19]。此外,还包含POU3F3基因,其编码的POU3F3 蛋白是真核转录因子,参与了大脑和肾脏的发育过程,致病性POU3F3变异导致Snijders Blok-Fisher 综合征,为常染色体显性遗传。ClinGen 报道的10 例包含该基因CNVs 的病例中,有8 例为重复致病,出现Snijders Blok-Fisher 综合征相关的智力障碍、全面发育迟缓、耳廓畸形、孤独症及其他精神行为异常等临床表现[20]。CKAP2L基因的致病变异可引起一种罕见的常染色体隐性颅指综合征,即Filippi 综合征,其特征是小头畸形、并指、身材矮小、智力残疾和畸形面部特征。目前仅有4 例包含该基因CNVs 患者信息,其中3 例为重复致病[21]。目前没有足够的证据确定上述基因AFF3、POU3F3、CKAP2L为3 倍剂量敏感基因,但是它们的重复在相当一部分病例中呈现3 倍剂量敏感导致的临床表型,与本例患儿高度一致,这些基因作为主要驱动因素,可导致患儿具有以智力障碍为主伴有生长迟缓、神经精神缺陷、多发畸形的复杂临床表型。而位于患儿重复片段中的TMEM127基因[22],为遗传性嗜铬细胞瘤的易感基因,目前已有足够证据表明该基因为单倍剂量敏感,因此可以解释为何患儿存在该基因的重复却无相关临床表型。明确该基因的变异方式并结合其单倍剂量敏感的致病特性,医生可以在遗传咨询和随访过程中,对患儿嗜铬细胞瘤患病概率做出科学预判。
2 号染色体p11.1-q21.1 区域的大片段重复,是引起患儿以智力障碍为主的多系统发育异常临床表型的遗传学病因。由于染色体重复或缺失的外显率不同,该区域包含的众多基因与疾病的关联程度、剂量敏感特性以及表型驱动优先级别不同,可能是导致患儿复杂特殊临床表型的潜在发病机制[23-24]。因本例患儿父母外周血染色体核型均为正常,提示患儿是新发突变,但不能除外父母存在生殖细胞嵌合的可能性。因此,在遗传咨询过程中,医生需要重视患儿父母的再生育诉求,建议对其进行有针对性的产前检查。
