异基因嵌合抗原受体T细胞技术的研究进展
2024-01-05赵茜,李锋
赵 茜,李 锋
东部战区总医院血液科,南京 210002
近十年来嵌合抗原受体T(chimeric antigen receptor T,CAR-T)细胞疗法在血液系统肿瘤中的应用和实践极大证明了免疫系统对抗肿瘤的有效性。嵌合抗原受体(chimeric antigen receptor,CAR)在经基因工程技术导入T细胞后,可与肿瘤相关抗原特异性结合,并通过信号转导激活T细胞,发挥抗肿瘤作用。截至2022年2月,全球已批准7款CAR-T细胞产品用于治疗非霍奇金淋巴瘤、急性B淋巴细胞白血病、多发性骨髓瘤[1-2]。目前,所有注册的治疗和大多数的临床试验都是基于二代的CAR,包括了一个细胞外抗原结合域(通常是抗体的单链可变片段)、一个T细胞受体(T cell receptor,TCR)的CD3ζ链和一个可与CD3ζ融合的共刺激域(CD28或CD137,也称为4-1BB)[3]。
虽然自体CAR-T细胞治疗拥有不错的临床数据,但由于它存在个性化定制成本过高、制备周期过长、细胞数量和扩增能力不足等问题,导致实用性下降,同时因为肿瘤微环境的多种免疫抑制机制可引起T细胞功能障碍,且随着肿瘤不断复发进展,治疗线数越多,临床治疗效果越差,所以肿瘤免疫微环境和治疗线数都会影响T细胞的生物学特性,从而导致自体T细胞在部分患者中无效[4]。目前研究人员使用健康供体的细胞制备异基因通用型CAR-T细胞,可通过优化制备工艺、质量控制,实现基因CAR-T细胞的规模化生产,不仅降低了成本,还能让更多患者获得治疗。
自2015年第1例异基因的CAR-T细胞应用于1岁急性淋巴细胞白血病患儿并获得分子生物学缓解以来,相继有患者成功接受异基因CAR-T细胞的治疗并序贯造血干细胞移植,这些成功的临床案例标志着异基因CAR-T细胞技术逐渐实现了从实验研究到临床应用的目标[5-6]。目前异基因方式主要存在两个问题:(1)异基因CAR-T细胞可能导致危及生命的移植物抗宿主病(graft-versus-host disease,GVHD);(2)异基因CAR-T细胞易被宿主免疫系统迅速消除,限制了它的抗肿瘤活性。下面我们将异基因CAR-T细胞制备的来源、如何改进制备流程、减少GVHD的产生和增加其抗肿瘤活性等研究进展做一综述。
异基因CAR-T细胞的来源
目前异基因CAR-T细胞制备的T细胞主要源于外周血单个核细胞(peripheral blood mononuclear cells,PBMC),如果发生HLA配型困难情况,常用来自脐带血的T细胞,也能用可再生的干细胞,如诱导多能干细胞(induced pluripotent stem cells,iPSC)。
健康供体来源选择健康供体来源的PBMC制备异基因CAR-T细胞,优势在于一方面制备成功的此类异基因CAR-T细胞是由不受肿瘤免疫效应影响或化学暴露损害的免疫细胞产生的,另一方面,异基因CAR-T细胞还能形成表达不同亚型的人类白细胞抗原(human leukocyte antigen,HLA)复合物的细胞库,以方便选择与患者HLA类型相匹配,根据HLA配型选择供体可能是降低细胞终产物异质性的关键因素。
脐带血来源使用脐带血来源的T细胞,具有独特抗原幼稚状态,对HLA相合位点的数量限制没有那么严格,且脐带血T细胞还可通过抑制活化T细胞核因子信号传导,减弱脐带血T细胞对受体抗原的同种反应性,从而降低GVHD的发生率和严重程度[7-8]。有报道胎盘干细胞可产生T细胞或自然杀伤(natural killer,NK)细胞,细胞同质性更好[9]。
干细胞来源iPSC具有无限的自我更新能力,可生成具有共同HLA单倍型的iPSC库,减少CAR-iPSC T细胞的异体排斥风险,且在HLA错配的情况下,还可利用基因编辑技术改造TCR,有效避免GVHD。使用iPSC来源的优势在于由多能细胞系产生的CAR-T细胞是同质的,但该方法的安全性和有效性尚需进一步临床验证。
降低GVHD的方法
因为αβT细胞上的TCR可识别主要组织相容性复合体(major histocompatibility complex,MHC)分子所呈递的多肽,从而引发GVHD,而GVHD又是引起异基因干细胞移植(allogeneic stem cell transplantation,Allo-SCT)患者死亡的主要原因,已有研究表明αβT细胞是急慢性GVHD发生发展的重要因素[10]。下面介绍几种异体CAR-T细胞降低GVHD的方法,包括使用供者来源的异体干细胞移植T细胞,使用病毒特异性记忆T细胞,使用非-αβT细胞和基于基因编辑构建TCR缺失的αβT细胞。
使用来自干细胞移植供体的异体CAR-T细胞此方式仅限于那些接受过Allo-SCT后复发的患者,CAR-T细胞可来自原始供体。供体来源的CAR-T细胞可纳入Allo-SCT的策略,以增强移植物抗肿瘤效应,而不增加GVHD的风险。Brudno等[11]研究表明20例复发的B细胞恶性肿瘤患者未化疗直接接受来自原始供体的CD19 CAR-T细胞,6例患者获得完全缓解,2例患者获得部分缓解,无一例发生GVHD。更多临床研究发现患者血液中CAR-T细胞的峰值早期可保持高水平,但CAR-T细胞在3周后无法维持在较高水平,长期效果尚需观察,这与之前报道是一致的[12-13]。
使用病毒特异性的记忆T细胞研究发现在Allo-SCT中纯化的病毒特异性记忆T细胞可以在不诱发GVHD的情况下预防病毒性疾病[14]。纯化的病毒特异性记忆T细胞有效降低了T细胞数量,减少了TCR的多样性,所以病毒特异性记忆T细胞中的TCR功能受限减低了GVHD的发生率。2022年12月美国血液学年会上公布了一项CD30 CAR修饰的Epstein-Barr病毒特异性T细胞疗法的临床I期数据,结果显示:14例晚期的淋巴瘤在3个不同剂量给药治疗后的总体缓解率为69.2%,完全缓解率为43%,安全性和耐受性良好[15]。
使用非αβ T细胞这种方法是完全避免使用αβT细胞,并设计另一种细胞类型来携带CAR。NK细胞是生物体天然肿瘤免疫监视的一部分,具有杀死肿瘤细胞的能力,不需要通过MHC途径激活,可降低同种反应风险,且CAR-NK细胞的制备可以使用现有的NK92细胞系,无需自体NK细胞,更加方便[16]。CAR-NK细胞疗法的另一个优势在于较少发生免疫介导毒性,如细胞因子释放综合征和神经毒性并发症等。一项11例患者接受来自脐带血的HLA不匹配抗CD19CAR-NK细胞治疗淋巴瘤的Ⅰ/Ⅱ期临床试验结果显示,无一例出现GVHD、细胞因子释放综合征或神经毒性并发症[17]。另有研究表明若在CAR-NK细胞扩增后同时表达白细胞介素(IL)-15,与CD19 CAR-NK细胞对照组相比,使用CD19 CAR-IL-15转导的NK细胞的淋巴瘤移植小鼠模型存活时间更长[18]。目前已有20多项关于CAR-NK细胞的Ⅰ/Ⅱ期临床试验正在进行,不过NK细胞的平均寿命有限,一般约为2周,这意味着可能需要反复输注CAR-NK细胞才能取得长期效果。
自然杀伤T(natural killer T,NKT)细胞属于T淋巴细胞,细胞表面表达NK标志物,恒定NKT细胞(invariant NKT,iNKT)是其主要亚型,表达一种高度限制性TCR,可通过识别CD1d所呈递的糖脂抗原转化为成熟的iNKT。与T细胞相比,iNKT表达更多的趋化因子受体(chemokine receptor,CCR),比如CCR1、CCR2、CCR3、CCR4等,可促进iNKT细胞浸润到肿瘤中去招募更多的免疫细胞进行杀伤,更重要的是iNKT细胞可以通过CD1d依赖的方式杀伤肿瘤相关巨噬细胞,并消除髓源性抑制细胞的免疫抑制活性[19]。目前认为供体iNKT细胞在Allo-SCT中可保护机体免受急性GVHD。临床研究发现,肿瘤中浸润的iNKT细胞的数量和比例与患者的总生存率呈正相关[20],但其实iNKT细胞数量较少,且在肿瘤中的浸润是不足的,同时多数肿瘤细胞为CD1d阴性或CD1d表达在肿瘤发展过程中下调,这使iNKT细胞无法直接识别,需要增加额外的共刺激信号。二代CAR构建的共刺激结构域中CD28或4-1BB与CD3ζ串联使用最广泛,研究表明4-1BB可提供有效的协同刺激以增强iNKT细胞的扩增能力和抗肿瘤效应,但同时会激活诱导CAR-iNKT细胞的细胞死亡,而包含CD28共刺激域的CAR-iNKT细胞却显示出抗凋亡的优势[21-22]。
候选细胞是γδ T细胞对肿瘤细胞有天然的细胞毒性反应。γδ T细胞仅占循环淋巴细胞的1%~5%,但在一些上皮部位占主导地位,如肠道、生殖器官、舌头和皮肤。γδ T细胞相比于αβ T细胞有如下优势:(1)在识别方式上,γδ T细胞以独立于MHC的方式识别其靶细胞,从而减少同种异体反应和GVHD的风险,是优质的异体细胞产品的来源。(2)γδ T细胞浸润在各种各样的组织中,可以快速响应靶标并释放效应细胞因子,特别是vδ1亚型,具有优于αβ T细胞的归巢优势,能更好地在肿瘤低氧环境中浸润和发挥功能。(3)γδ T细胞对于肿瘤的识别和杀伤不依赖于单一抗原的表达,相反,它们通过在细胞膜上表达的各种先天细胞毒性受体来识别各种肿瘤细胞上的多种抗原,扩大了可用于杀死肿瘤细胞的靶点范围,减少了单抗原丢失导致肿瘤免疫逃逸的机会。
目前γδCAR-T细胞已在白血病、肝癌的临床前模型中显示出不错的抗肿瘤效应[23-24],但体外扩增培养工艺和细胞的工程化改造仍面临挑战,且其疗效与普通CAR-T细胞相比也是有限的。
使用基因编辑技术目前主要依赖锌指核酸酶、转录激活子样效应因子核酸酶和规律性间隔的短回文序列重复簇(clustered regularly interspaced short palindromic repeat,CRISPR)/CRISPR相关基因9(CRISPR-associated genes 9,Cas9)等平台来进行基因编辑[25]。在基因组的TCR和HLA的位点进行特异性双链断裂,通过非同源末端连接或同源定向重组方式进行修复,从而实现敲除TCR和HLA来减弱或消除GVHD并提高CAR-T的存活能力;或者同时敲除T细胞抑制性受体的基因,如程序性死亡受体-1、细胞毒T淋巴细胞相关抗原4、T细胞免疫球蛋白黏蛋白-3等,减少T细胞耗竭,增强CAR-T的杀伤功能。总的来说锌指核酸酶特异性高,脱靶率低,但细胞毒性大,成本高,而CRISPR/Cas9由于其设计简单,操作方便,成本较低,编辑高效,可实现多靶点编辑,目前在进行广泛开发,但是它的脱靶率较高。以上技术目前已在多个临床试验中使用(表1),包括靶向CD19的UCART019(NCT03166878)、靶向CD70的CTX130(NCT04502446、NCT04438083)、靶向B细胞成熟抗原的CTX120(NCT04244656)和靶向CD5的CT125A(NCT04767308)等。
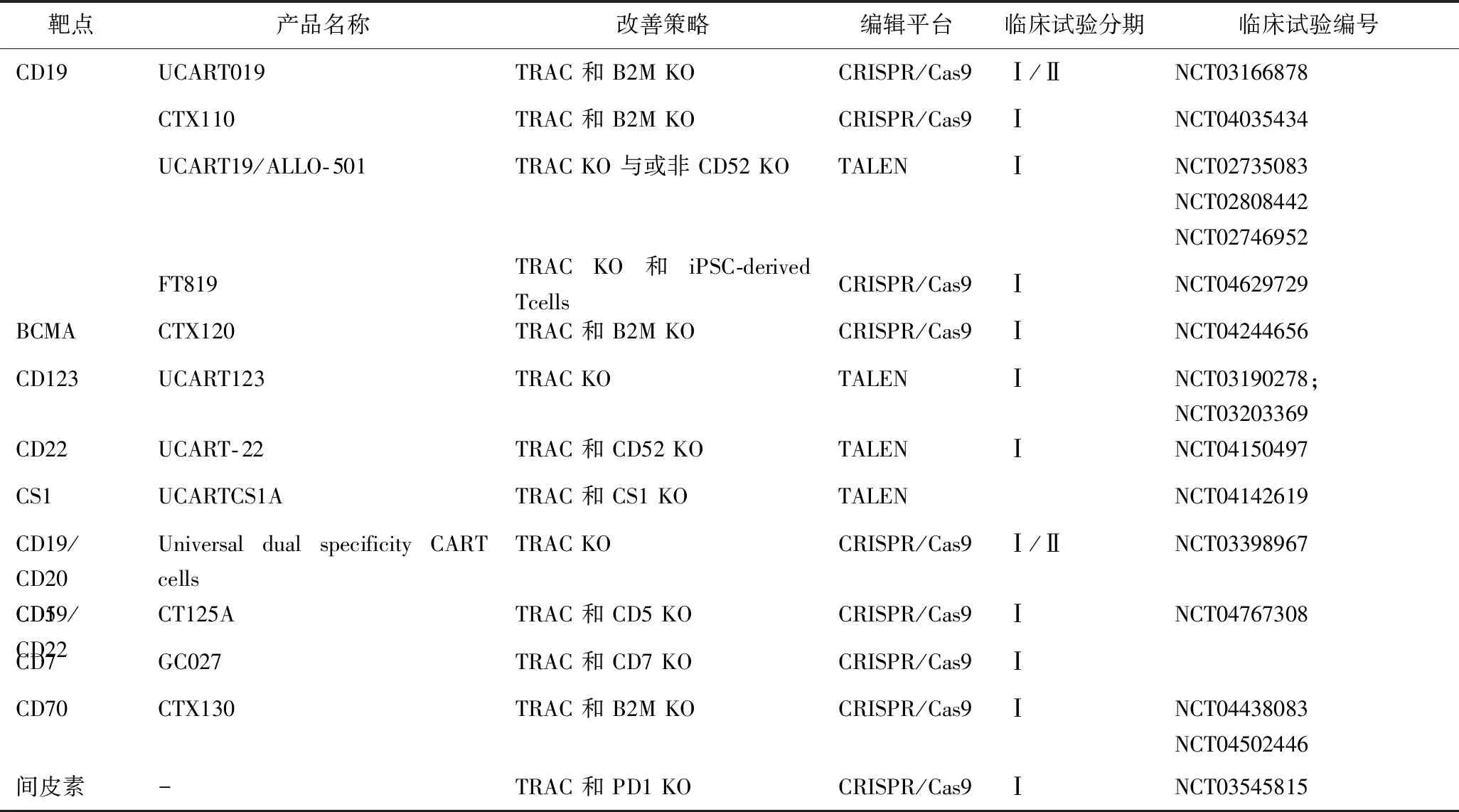
表1 基于基因编辑的异基因CAR-T临床试验
基于目前越来越多的临床实践经验,发现输注前先进行去除T淋巴细胞的预处理化疗方案,会更有利于体内CAR-T细胞的扩增,常用的去除T淋巴细胞的预处理化疗方案为氟达拉滨与环磷酰胺。在异基因CAR-T治疗中为了更彻底的淋巴细胞耗竭,T细胞被设计成敲除编码T细胞受体ɑ恒定链(T-cell receptor alpha constant chain,TRAC)和CD52基因的结构,从而获得了TCR缺失和对抗CD52单克隆抗体阿仑单抗产生抗药性,该单抗可用于消除宿主T细胞(表达CD52)并避免异体排斥。在预处理中添加阿仑单抗可以进一步抑制宿主的同种异体免疫排斥反应,并通过扩增异基因CAR-T细胞来延长其治疗窗口期。在2项复发儿童和成人急性B淋巴细胞白血病的I期临床试验中发现,经异基因CD19CAR-T细胞治疗后所有获得完全缓解的患者的预处理方案为氟达拉滨与环磷酰胺+阿伦单抗(14/21,67%),4例没用阿伦单抗的患者未见CD19CAR-T扩增或抗白血病效应[26]。一项针对多发性骨髓瘤的敲除TRAC和CD52的异基因B细胞成熟抗原CAR-T临床试验显示治疗后总体缓解率为60%,非常好的部分缓解率为40%,45%的患者出现细胞因子释放综合征,无患者出现神经毒性;扩增高峰在7 d左右,并可持续120 d,未观察到GVHD的发生[27]。
CD7也是其敲除靶点,有研究表明敲除TRAC和CD7的异基因CAR-T在体外和免疫缺陷小鼠的细胞系和原发性急性T淋巴细胞白血病细胞中保持了强大的抗白血病作用,未发现GVHD[28];而且近期一项由CRISPR编辑的TCR和CD7的异基因CAR-T(GC027)的临床试验显示2例强化疗后难治性/复发性T-ALL患者在单次输注GC027后获得完全缓解,所检测到的微小残留病灶都是阴性[29]。
CD4+T细胞介导同种免疫排斥反应,表达可被调节因子X锚蛋白重复序列调节蛋白质和Ⅱ类MHC反式激活子调节[30]。研究发现与异基因PBMC共培养的三重敲除β2微球蛋白、MHC反式激活子和TRAC的异体抗CD19 CAR-T细胞比双重敲除TRAC和β2微球蛋白表现出更好的持久性[31]。iPSC中也有类似的基因编辑,比如敲除β2微球蛋白、MHC反式激活子和CD155(编码NK细胞的活化配体)以及转导HLA-E,这些低免疫原性细胞将大大减少来自CD8+T细胞、CD4+T细胞和NK细胞带来的免疫反应并维持其抗肿瘤毒性[32]。
总 结
总而言之,自体CAR-T细胞疗法极大地改善了血液系统肿瘤患者的预后,逐渐成为抗肿瘤治疗中的明星产品,但价格昂贵、制备周期和质控问题限制了自体CAR-T细胞疗法广泛应用。异基因CAR-T细胞选择不同来源的T细胞制备,并可利用多种基因编辑技术来减少GVHD和增强杀伤力,极大提高了异基因CAR-T细胞疗法的可及性和适用性。但异基因CAR-T细胞技术才刚刚起步,仍然面对诸多挑战,如异基因CAR-T细胞疗法中细胞的扩增、持久性问题,脱靶效应等,随着对免疫系统作用机制研究的深入、基因编辑技术的进步和CAR-T结构的不断优化,相信异基因CAR-T会像血制品一样成为一种常规的活性药物,给血液系统肿瘤患者提供安全高效和更高性价比的方案,带来更光明的治疗前景。
