薄层鉴别结合多成分测定方法评价苓桂术甘颗粒质量
2024-01-03马晓敏周建新肖苏萍谢彬李龙妹陈蒙杜杰王继永卢亚楠
马晓敏,周建新,肖苏萍*,谢彬,3,李龙妹,3,陈蒙,杜杰,王继永,卢亚楠
1.中国中药有限公司,北京 102600;2.北京城市学院,北京 100191;3.江西中医药大学,江西 南昌 330004
苓桂术甘汤出自东汉·张仲景《金匮要略》,主治心下痰饮、胸胁支满、三焦有水气,温阳以化饮,使水饮下行,疗效确切,是苓桂剂的代表方剂。原方由茯苓、桂枝、白术和甘草4 味药组成,“以水六升,煮取三升,分温三服”[1]。苓桂术甘汤是治疗痰饮的代表方剂,痰饮疾病症状复杂,易累及其他系统。现代临床研究表明,苓桂术甘汤对于心系疾病、肺系疾病和肾系疾病等均有确切疗效[2-3]。桂枝的主要有效成分为桂皮醛、肉桂酸、原儿茶酸和香豆精等,对中枢神经系统具有镇静、镇痛、解热和抗惊厥作用,桂皮醛、肉桂酸为其中主要有效成分。白术中主要含有多糖、挥发油、内酯类和绿原酸类成分。绿原酸类成分具有抑菌、抗感染、抗肿瘤、调血脂、调血糖、保护心血管、保护神经系统的作用,其在抑菌、抗感染等方面研究较多,并可与其他有效成分共同发挥药理活性[4-7]。苓桂术甘颗粒中白术内酯类成分含量较低,且采用高效液相色谱法较难分离。结合近年来相关研究[4-7],选择肉桂酸、桂皮醛、新绿原酸、绿原酸和隐绿原酸作为定量指标,肉桂酸、白术内酯Ⅱ、白术内酯Ⅲ作为定性指标评价苓桂术甘颗粒的质量。
依据《按古代经典名方目录管理的中药复方制剂药学研究技术指导原则(试行)》[8]的要求,将苓桂术甘汤经现代制备工艺制成使用便携、剂量准确的颗粒制剂。经典名方研发需“遵古”,即按照古籍记载的工艺确定基准样品标准后,颗粒制剂需与基准样品质量保持一致,并建立制剂的相关质量控制方法。目前,对于经典名方苓桂术甘汤质量标准的研究多针对基准样品进行,尚未有公布的苓桂术甘颗粒质量标准。本研究通过薄层鉴别及含量测定方法对苓桂术甘颗粒的质量进行评价,以期为苓桂术甘颗粒的质量控制提供参考。
1 材料
1.1 仪器
Visualizer 型薄层色谱数码成像系统、Linomat型薄层色谱半自动点样仪(卡玛公司);AE 240 型电子天平[梅特勒托利多仪器(上海)有限公司];XB320M 型电子天平(Presisa 公司);KQ-500E 型超声波清洗仪(昆山市超声仪器有限公司);3-16L 型离心机(Sartorius 公司);e2695 型高效液相色谱仪(Waters公司)。
1.2 试药
苓桂术甘颗粒(自制,批号分别为LG2220411001、LG220331003、LG220331004);对照药材桂枝、白术(批号分别为121191-201906、120925-202013),对照品肉桂酸、桂皮醛、白术内酯Ⅱ、白术内酯Ⅲ、绿原酸(批号分别为110786-201604、110710-202223、111976-201501、111978-201501、110753-202119,纯度分别为99.8%、99.5%、96.3%、≥98%、≥98%)均购于中国食品药品检定研究院;苓桂术甘桂枝、白术阴性颗粒(自制,批号分别为LG220715GZY、LG220715BZY);对照品新绿原酸、隐绿原酸(成都埃法生物有限公司,批号分别为AF20030804、AF21010310,纯度分别均为98%);水为娃哈哈纯净水;甲醇、乙酸乙酯、石油醚、甲酸、乙醚均为分析纯;乙腈、磷酸为色谱纯;硅胶GF254薄层板(青岛谱科分离材料有限公司)。
2 方法与结果
2.1 薄层色谱鉴别
2.1.1 对照品溶液制备 精密称定桂皮醛17.31 mg、肉桂酸10.69 mg、白术内酯Ⅱ5.88 mg、白术内酯Ⅲ9.32 mg,分别置于10 mL 量瓶中甲醇定容,得桂皮醛、肉桂酸、白术内酯Ⅱ、白术内酯Ⅲ质量浓度分别为1.731、1.069、0.588、0.932 mg·mL-1的对照品溶液。
2.1.2 对照药材溶液制备 桂枝对照药材:取桂枝对照药材粉末2 g,加乙醚10 mL,浸泡30 min,时时振摇,滤过,滤液挥干,残渣加三氯甲烷1 mL 使溶解,作为对照药材溶液。
白术对照药材:取白术对照药材粉末0.5 g,加正己烷2 mL,超声处理15 min(250 W,40 kHz),滤过,取滤液作为对照药材溶液。
2.1.3 供试品溶液制备 取苓桂术甘颗粒5 g,加水20 mL 使溶解,乙醚萃取2 次,每次20 mL。收集乙醚层,挥干,残渣加甲醇1 mL 复溶,作为供试品溶液。
2.1.4 阴性样品溶液制备 取缺白术的苓桂术甘颗粒和缺桂枝的苓桂术甘颗粒各5 g,加水20 mL 使溶解,乙醚萃取2 次,每次20 mL。收集乙醚层,挥干,残渣加甲醇1 mL复溶,作为阴性样品溶液。
2.1.5 薄层色谱鉴别 照薄层色谱法试验,吸取桂皮醛对照品溶液、肉桂酸对照品溶液各5 μL,白术内酯Ⅱ、白术内酯Ⅲ对照品溶液各5 μL,桂枝对照药材溶液5 μL,白术对照药材溶液8 L,供试品溶液20 μL,白术、桂枝阴性样品溶液各5 μL,分别点于同一硅胶GF254薄层板上,以石油醚-乙酸乙酯-甲酸(17∶4∶0.1)为展开剂,预先饱和15 min,展开,取出,晾干,置于紫外254 nm 检视,在与桂皮醛对照品、肉桂酸对照品、桂枝对照药材色谱相应的位置上,显相同颜色斑点。喷以5%二甲氨基苯甲醛的10%硫酸乙醇溶液,在105 ℃下烘烤3 min,置于紫外366 nm检视,在与白术内酯Ⅱ对照品、白术内酯Ⅲ对照品、白术对照药材色谱相应的位置上,显相同颜色斑点(图1)。

图1 苓桂术甘颗粒薄层鉴别
2.1.6 3 批苓桂术甘颗粒薄层色谱鉴别 对3 批实验室自制苓桂术甘颗粒样品使用2.1.5 项下方法进行薄层鉴别,见图2。

图2 苓桂术甘颗粒薄层鉴别多批验证
2.2 含量测定
2.2.1 色谱条件 X-Bridge C18色谱柱(250 mm×4.6 mm,5 μm);以乙腈(A)-0.1%磷酸水溶液(B)为流动相洗脱梯度(0~25 min,7%~13%A;25~30 min,13%~30%A;30~40 min,30%A;40~50 min,30%~32%A);流速:1 mL·min-1;柱温:30 ℃;检测波长分别为290、325 nm;进样量:10 μL,色谱图见图3。

图3 苓桂术甘颗粒高效液相色谱图
2.2.2 对照品溶液的制备 精密称取对照品新绿原酸3.150 mg、绿原酸2.720 mg、隐绿原酸3.185 mg、肉桂酸4.735 mg、桂皮醛5.06.mg,置10 mL量瓶中,吸取该对照品母液1 mL至50 mL量瓶甲醇定容,即得新绿原酸、绿原酸、隐绿原酸、肉桂酸、桂皮醛质量浓度分别为6.30、5.44、6.37、9.47、10.12 μg·mL-1的对照品溶液。
2.2.3 供试品溶液的制备 称取苓桂术甘颗粒约1 g,精密称定,置50 mL 锥形瓶中,精密加入50%甲醇20 mL,称定质量,超声处理(250 W,45 kHz)30 min,放至室温,用甲醇补足质量,摇匀,滤过,取续滤液,即得。
2.2.4 专属性考察 按2.2.3 项下方法制备苓桂术甘颗粒样品溶液、阴性样品溶液、阳性样品溶液,按2.2.1 项下色谱条件进样,观察阴性样品在目标峰相同位置是否有干扰,结果见图4。桂枝、白术阴性样品无干扰,此方法专属性强。

图4 苓桂术甘颗粒缺桂枝、白术阴性图谱
2.2.5 线性关系考察 采用二倍稀释法对混合对照品母液进行稀释,新绿原酸质量浓度分别为68.40、34.20、17.10、7.88、3.90、0.95 μg·mL-1;绿原酸质量浓度分别为54.60、27.30、13.65、6.83、3.41、1.71 μg·mL-1;隐绿原酸质量浓度分别 为63.70、31.85、15.93、7.96、3.90、0.98 μg·mL-1;肉桂酸质量浓度分别为10.12、5.06、1.27、0.63、0.32、0.16 μg·mL-1。按2.2.1 项下色谱条件进样,以进样量为横坐标(X),峰面积为纵坐标(Y)绘制标准曲线,线性关系见表1。
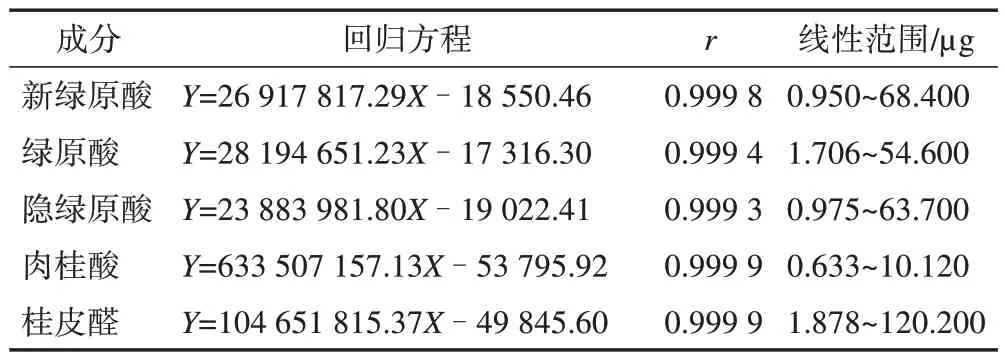
表1 苓桂术甘颗粒中5个成分线性关系
2.2.6 精密度试验 取供试品溶液,按2.2.1 项下色谱条件连续进样6 次,记录峰面积。结果显示,新绿原酸、绿原酸、隐绿原酸、肉桂酸和桂皮醛的峰面积的RSD 分别为2.87%、0.60%、0.78%、0.44%、0.62%,表明仪器精密度良好。
2.2.7 稳定性试验 取供试品溶液,按2.2.1 项下色谱条件分别在0、2、4、8、12、24 h 进样,记录峰面积。结果显示,新绿原酸、绿原酸、隐绿原酸、肉桂酸和桂皮醛峰面积的RSD 分别为1.62%、0.52%、2.03%、0.15%、0.49%,表明供试品溶液在24 h内稳定性良好。
2.2.8 重复性试验 取苓桂术甘颗粒,按2.2.3 项下方法平行制备6份供试品溶液,按2.2.1项下色谱条件进样,记录峰面积并计算含量。结果显示,新绿原酸、绿原酸、隐绿原酸、肉桂酸和桂皮醛的平均质量分数分别为0.050 0、0.207 8、0.061 7、0.223 6、1.205 6 mg·g-1,RSD 分别为2.14%、0.87%、1.12%、0.29%、1.00%,表明该测定方法重复性良好。
2.2.9 加样回收率试验 精密称取已知含量的苓桂术甘颗粒,按1∶1 比例加入混合对照品,平行制备6 份供试品溶液,按2.2.1项下色谱条件测定,计算回收率及RSD,结果新绿原酸、绿原酸、隐绿原酸、肉桂酸、桂皮醛的加样回收率分别为109.46%、93.45%、97.04%、103.70%、102.41%,RSD 均小于3%。
2.2.10 3 批苓桂术甘颗粒5 个成分的含量测定 结果见表2。
3 讨论
3.1 薄层鉴别条件考察
薄层鉴别色谱图中因颗粒剂中桂皮醛斑点较弱,考虑到桂皮醛成分不稳定,桂枝对照药材在366 nm下显示斑点与白术内酯类成分比移值接近;白术对照药材斑点较多,但主要斑点为白术内酯Ⅱ、白术内酯Ⅲ。因此,选择肉桂酸、白术内酯Ⅱ、白术内酯Ⅲ作为薄层鉴别指标鉴别苓桂术甘汤颗粒。考察了不同提取方式(回流提取、超声提取、有机溶剂萃取),3 种提取方式对肉桂酸斑点影响较大,采用有机溶剂萃取法获得的肉桂酸斑点最为清晰。因此,选择有机溶剂萃取。此外,考察了不同取样量、不同点样量,其中取样量5 g、供试品点样量15 μL,斑点最为清晰。不同温度对薄层条件无影响,但是湿度对薄层条件影响较大,湿度越大,薄层色谱分离越差,相对湿度为35%时效果较好。
3.2 含量测定条件考察
对含量测定的色谱条件和提取条件进行考察。考察了2 种型号色谱柱CAPCELL PAK、X-Bridge,乙腈-0.1%磷酸水溶液、甲醇-0.1%磷酸水溶液、乙腈-水对色谱图的影响。结果X-Bridge 色谱柱、乙腈-0.1%磷酸水溶液为流动相效果更佳;又分别考察了超声提取和回流提取,甲醇提取、乙醇提取和水提取,25%甲醇、50%甲醇、75%甲醇提取,提取15、30、45 min,加溶剂10、20、30 mL,结果以加25%甲醇20 mL提取30 min最佳。
3.3 方法可行性的探讨
《中华人民共和国药典》2020 年版收载的中药复方制剂中薄层鉴别或含量测定方法大多是多项鉴别、多项含量测定、制备多种供试品溶液、多块薄层板、展开多次、鉴别不同药材、不同色谱条件、测定不同成分的传统鉴别模式。为排除干扰,样品前处理复杂、繁琐、试剂耗材消耗大、检测时间长,而且对环境造成污染。本研究建立了苓桂术甘颗粒中桂枝、白术2 味中药同时定性、定量质量控制方法,可缩短检测时长、节约试剂耗材、减少环境污染等。由于桂枝、白术2 味中药的成分在苓桂术甘汤中的含量差异较大,颗粒制剂与基准样品因辅料等略有差异,本研究的含量测定方法从经济适用性和标准方法可行性上具有一定参考价值,但颗粒质量是否完全与基准样品一致需多批量样品验证。
4 结语
苓桂术甘颗粒为古代经典名方苓桂术甘汤经现代工艺制备而成的复方制剂,成分复杂,质量标准建立和控制难度较大。本研究建立的方法还未见报道。建立不同的质量控制方法对保证经典名方的质量具有一定意义。本研究使用了薄层鉴别和高效液相方法同时对苓桂术甘颗粒中的桂枝、白术相关成分进行测定,在短时间内同时进行2 味中药的薄层鉴别和含量测定,能够更好地反映苓桂术甘颗粒的信息,经方法学验证,方法可行。
