碳氧血红蛋白在新生儿Gilbert综合征中的鉴别诊断价值*
2024-01-03陈亚丽胡振红邱建武
陈亚丽,胡振红,邱建武,胡 丽
汕头大学医学院附属粤北人民医院新生儿科,广东韶关 512026
新生儿高胆红素血症是临床上常见的疾病,然而,导致高胆红素血症的病因是多种多样的,不同病因导致的高胆红素血症,临床结局也不同。血清总胆红素水平取决于胆红素产生和消除之间的平衡,影响胆红素产生和消除的因素发生改变都可以导致胆红素水平升高。在不明原因的病理性黄疸患儿中Gilbert综合征和葡萄糖-6-磷酸脱氢酶(G-6-PD)缺乏症是最常见的2种疾病[1]。
在胆红素的形成过程中会产生胆绿素、铁,同时产生等当量的一氧化碳(CO)[2],CO与血红蛋白结合形成碳氧血红蛋白(COHb),COHb是胆红素生成的敏感指标。
Gilbert 综合征是尿苷二磷酸葡萄糖醛酸基转移酶1A1(UGT1A1)基因变异导致的一种遗传代谢性疾病,也称为Meulengracht病、全身性肝功能障碍和家族性非溶血性黄疸,由于UGT1A1基因编码的UGTs同工酶1A1活性减低,造成肝细胞摄取非结合胆红素障碍,表现为高非结合胆红素血症,其特征是非结合(间接)胆红素水平轻度和间歇性升高[3]。G-6-PD缺乏症是由于G-6-PD活性降低或性质改变导致红细胞溶血,在新生儿期可表现为黄疸。
目前对Gilbert 综合征患儿进行COHb检测的研究报道很少,本文对Gilbert综合征患儿和G-6-PD缺乏症患儿的全血COHb进行对比分析,探讨COHb在新生儿Gilbert综合征和G-6-PD缺乏症患儿中的早期鉴别诊断价值。
1 资料与方法
1.1一般资料 选取本院新生儿科 2020 年11 月至2022年5月收治、确诊的30例新生儿Gilbert综合征患儿作为观察组。纳入标准:所有患儿均进行UGT1A1基因检测,基因变异为纯合变异或来源父母的复合杂合变异确诊为Gilbert综合征。排除标准:合并了其他可导致黄疸的疾病,如新生儿溶血病、头颅血肿、颅内出血、红细胞增多症等;未进行COHb检测。以同期收治的69例G-6-PD缺乏症患儿为对照组。本研究经本院医学伦理委员会审批通过;患儿家属同意并签署知情同意书。
1.2方法 两组患儿入院时采用含肝素钠作抗凝剂的一次性预充型动脉血气针(美国BD公司)采集0.5 mL动脉血,采用GEM Premier 4000全自动血气分析仪进行COHb检测,以COHb占总血红蛋白(tHb)的百分比(%)表示,并同时进行总胆红素(TBil)检测,由新生儿科医生按照仪器说明书进行操作,检测试剂和质控试剂品均为该仪器的配套试剂。另外收集两组患儿的性别、年龄一般资料情况。
1.3统计学处理 采用SPSS 22.0 软件进行数据分析。偏态分布的计量资料用M(P25,P75)表示,两组间比较采用轶和检验;计数资料以例数、百分率表示,组间比较采用χ2检验。以P<0.05为差异有统计学意义。
2 结 果
2.1两组患儿一般资料比较 两组患儿的性别、年龄情况比较见表1,对照组患儿的日龄大于观察组(P<0.05)。
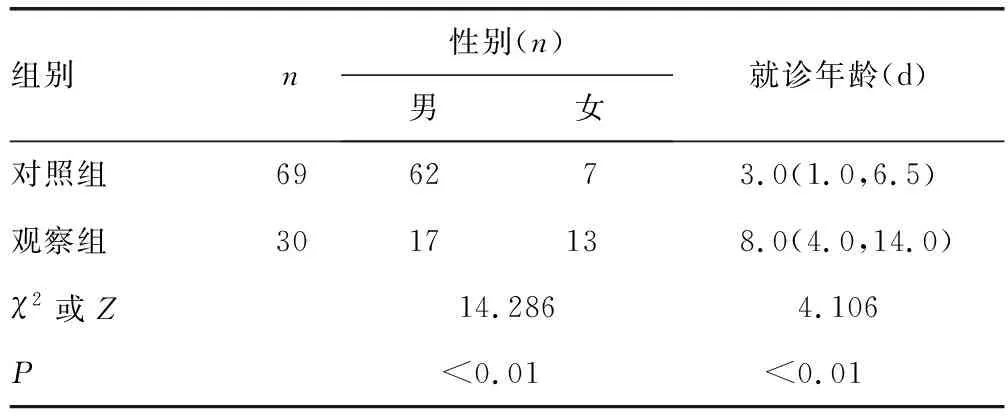
表1 两组患儿一般情况比较
2.2UGT1A1基因变异类型 30例Gilbert 综合征患儿UGT1A1基因位点变异情况见表2。

表2 Gilbert 综合征患儿 UGT1A1基因外显子变异情况
2.3两组患儿COHb、TBil水平比较 观察组患儿COHb水平明显低于对照组患儿,差异有统计学意义(P<0.05);而TBil水平在两组间比较,差异无统计学意义(P>0.05)。见表3。
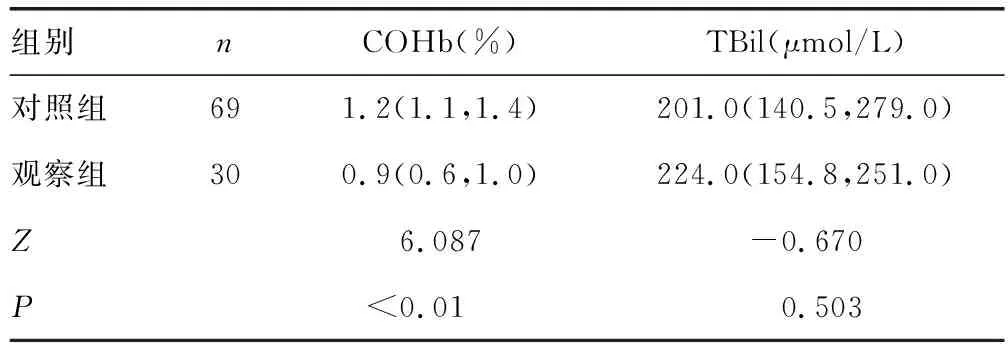
表3 两组患儿COHb、TBil水平比较 [M(P25,P75)]
3 讨 论
正常情况下,红细胞衰老后,在网状内皮系统内,血红蛋白被裂解产生珠蛋白和血红素,血红素在血红素加氧酶(HO)作用下通过氧化释放出铁,并产生CO,生成胆绿素,胆绿素再通过胆绿素还原酶还原为非结合胆红素,这种亲脂性分子扩散到血液中并与血液循环中的清蛋白结合,运送到肝脏内,胆红素进入肝细胞中,通过UGT1A1进行葡萄糖醛酸化,生成结合型胆红素,再通过多药耐药蛋白-2(MRP2)从胆小管排泄进入胆道系统,并最终通过尿液、粪便排出体外[4]。
UGT1A1是肝脏内清除胆红素的唯一限速酶[5],如果编码UGT1A1的基因变异,导致UGT1A1活性不同程度下降,使得非结合胆红素不能有效转化为结合胆红素,从而引起血清非结合胆红素水平升高,出现新生儿高胆红素血症的临床表现。由于UGT1A1的基因变异位点不同,可导致UGT1A1活性降低程度不一样,呈现出3种临床表型,如果UGT1A1酶活性完全消失,临床上表现为Crigler-Najjar SyndromeⅠ(CN-Ⅰ)型,如果UGT1A1酶的活性降低到正常值的10%以下,则临床表现为Crigler-Najjar Syndrome Ⅱ(CN-Ⅱ)型,Gilbert综合征患者肝细胞中UGT1A1活性降低到正常值的30%以下[6]。
目前对新生儿Gilbert 综合征的研究较少,成人Gilbert 综合征诊断方法有肝活检、饥饿试验、利福平试验、鲁米那治疗试验等,但这些方法均不适合健康体检人群及新生儿特殊人群,这部分人群诊断Gilbert 综合征的金标准就是检测到UGT1A1基因变异[7]。全球范围内已知UGT1A1基因变异类型有130余种,在不同的地区、种族、个体间有较大差异。既往研究显示我国UGT1A1基因c.211G>A变异频率高[8],这与本研究结果是一致的,UGT1A1基因c.211G>A变异是新生儿病理性黄疸发生的高危因素。有研究发现c.211G>A(p.Gly71Arg)与c.1456T>G(p.Tyr486Asp)复合杂合变异可表现为CN-Ⅱ型[6],但在本研究中,2例c.211G>A与c.1456T>G复合杂合变异者并未出现典型的CN-Ⅱ型表现,这需要扩大样本量进一步研究。
G-6-PD缺乏症是已知最常见的酶缺乏症之一,在我国南方多见。G-6-PD通过还原型谷胱甘肽保护细胞免受氧化损伤,在G-6-PD缺乏的情况下,还原型谷胱甘肽不能生成,氧化反应不能被消除,红细胞发生氧化损伤,造成细胞膜受损和溶血[9]。G-6-PD缺乏症以X连锁不完全显性的方式遗传,男性患者G-6-PD活性明显低于女性,在临床上男性患儿多于女性患儿。有人注意到,与女性相比,男性Gilbert综合征患者的临床表现会更明显,这可能与男性胆红素负荷较高,性激素对UDP葡萄糖醛酸基转移酶的影响有关[3],但在新生儿Gilbert综合征中,性别的差异不明显。
评估新生儿溶血最准确的方法是评估内源性CO的产生,从血红素产生的每个胆红素分子,伴随释放一个CO分子。测定血液中的COHb及呼气末CO(ETcO)都可以间接反映体内胆红素的产量及红细胞破坏程度,但由于新生儿为特殊群体,无法主动配合,在临床应用ETcO存在一定局限[10]。血气分析仪是每个新生儿重症监护室(NICU)常用的床旁仪器,大多数的血气分析仪都有测量COHb的功能,因此COHb检测可以轻松服务于临床,而无须昂贵的实验室测试及额外的静脉穿刺抽血或额外使用测量呼出二氧化碳的设备[2]。GEM Premier 4000血气分析仪在临床上被广泛应用,0.5 mL全血就能快速定量检测COHb、TBil等指标,能自动进行质量控制和校准,相对方便、快捷,常用于标本急诊检测及床旁检测[11]。
本研究发现Gilbert综合征患儿就诊年龄晚于G-6-PD缺乏症患儿,原因可能是Gilbert综合征患儿很多是因为黄疸延迟消退、黄疸反复而来就诊,这样造成观察组研究队列就诊年龄过大。本研究中Gilbert综合征患儿COHb水平明显低于对照组,而两组间TBil水平无差异,原因可能是COHb水平升高与TBil并不是同步的,COHb水平升高要早于TBil水平的升高[2];COHb可反映溶血的严重程度,而Gilbert综合征引起TBil水平升高,是由于胆红素代谢限速酶异常导致清除胆红素能力下降,从而表现出病理性黄疸。
COHb检测具有操作简单、采血量少、报告快速、可床旁操作等优点,亦可作为新生儿Gilbert 综合征的鉴别诊断参考指标。对于那些原因不明、COHb水平不高的新生儿高胆红素血症患儿,要考虑Gilbert综合征的可能,尽快完善基因检测。
