2-硝基-1H-咪唑甲酸乙酯衍生物的合成
2024-01-02马鸿钊陈夏彬李正义
王 顺, 马鸿钊, 陈夏彬, 石 靖, 杨 科, 李正义*
(1. 常州大学 石油化工学院 江苏省绿色催化材料与技术重点实验室,江苏 常州 213164;2. 江苏华景分子影像与药物研究院有限公司,江苏 常熟 215500)
1-甲基-2-硝基-1H-咪唑-5-甲酸乙酯是一种重要的医药中间体,可用于合成药物TH-302[1]和ICF05016[2]。TH-302是一种具有高细胞毒性的选择性低氧激活型DNA烷化剂,可用于治疗多发性骨髓瘤和人乳头瘤病毒阴性头颈鳞状细胞癌[3-4]; ICF05016是一种具有QA(季铵盐)功能的磷酰胺芥子[5],可用于治疗软骨肉瘤[6]。
CAVALLERI等[7]首次报道了1-甲基-2-硝基-1H-咪唑-5-甲酸乙酯的合成方法,以3,3-二乙氧基-2-(甲基氨基)丙酸乙酯为原料与氨基氰缩合环化后氧化氨基得到最终产品,然而该方法原料制备繁琐,不适于工业化生产。传统的工业制备方法如图1所示,首先MATTEUCCI等[8]以肌氨酸甲酯盐酸盐为初始原料,经缩合、环化和氧化等过程实现了1-甲基-2-硝基-1H-咪唑-5-甲酸乙酯的大规模合成;随后姚坤等[9]在环化步骤中通过降低温度提高了反应的选择性和收率,且在目标产品的纯化中用丙酮重结晶代替硅胶色谱柱提纯,降低了成本和实验繁琐程度;最后O’CONNOR等[10-11]以溶解性能更佳的肌氨酸乙酯盐酸盐代替肌氨酸甲酯盐酸盐,以四氢呋喃和甲酸乙酯作共溶剂,进一步提升了反应的效率,且在氨基氧化步骤中通过降低水的比例进一步提高了目标产品的收率。但这些方法仍存在很大的局限性,即以肌氨酸酯盐酸盐为初始原料会导致合成的1-甲基-2-硝基-1H-咪唑-5-甲酸乙酯的氮原子只能被甲基取代,不利于后续的各种药物分子的结构修饰。

图1 1-甲基-2-硝基-1H-咪唑-5-甲酸乙酯的传统合成路线Figure 1 Traditional synthesis route of ethyl 1-methyl-2-nitro-1H-imidazole-5-carboxylate
本文设计了一条新的合成路线(图2),以2-氨基嘧啶(1)和3-溴丙酮酸乙酯(2)为原料,合成咪唑[1,2-a]嘧啶-3-甲酸乙酯(3)和咪唑[1,2-a]嘧啶-2-甲酸乙酯(4);然后在水合肼作用下开环生成2-氨基-1H-咪唑-5-甲酸乙酯(5);接着在亚硝酸钠和乙酸作用下生成2-硝基-1H-咪唑-5-甲酸乙酯(6);最后在碳酸钾作用下利用不同碘代试剂生成一系列1-烷基-2-硝基-1H-咪唑-5-甲酸乙酯(7a~7d)及其异构体1-烷基-2-硝基-1H-咪唑-4-甲酸乙酯(8a~8d)。
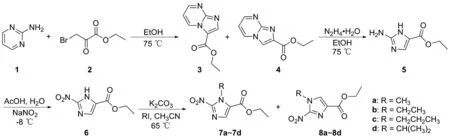
图2 1-烷基-2-硝基-1H-咪唑甲酸乙酯的合成路线Figure 2 Synthesis route of ethyl 1-alkyl-2-nitro-1H-imidazolecarboxylates
1 实验部分
1.1 仪器与试剂
AVANCE 300/400 MHz型核磁共振仪(CDCl3或DMSO-d6为溶剂,TMS为内标);LCMS-2020型液相质谱联用仪。
所用试剂均为分析纯。
1.2 合成
(1)3和4的合成[12]
在反应瓶中依次加入3-溴丙酮酸乙酯7.35 mL(52.60 mmol), 2-氨基嘧啶5.00 g(52.60 mmol)和乙醇(80 mL),升温至75 ℃搅拌反应16 h。冷却至室温,减压蒸除溶剂后加入二氯甲烷(30 mL)和饱和碳酸氢钠水溶液(60 mL),分液后再用饱和碳酸氢钠洗涤有机层2次,有机相经无水硫酸钠干燥,减压浓缩得到棕色固体,用乙酸乙酯洗涤并干燥得到4;滤液减压浓缩经硅胶柱层析(洗脱剂:石油醚 ∶乙酸乙酯=1 ∶1,V∶V)纯化得到3。
3:灰白色固体,收率20%, m.p.108~110 ℃(文献[13]报道熔点为109~110 ℃);1H NMR(DMSO-d6, 300 MHz)δ: 8.98(dd,J=6.90 Hz, 2.10 Hz, 1H), 8.68(dd,J=4.20 Hz, 2.10 Hz, 1H), 8.51(s, 1H), 7.17(dd,J=6.90 Hz, 4.20 Hz, 1H), 4.34(q,J=7.20 Hz, 2H), 1.34(t,J=7.20 Hz, 3H);13C NMR(DMSO-d6, 75 MHz)δ: 162.93, 153.51, 147.88, 136.61, 136.35, 117.09, 110.59, 60.93, 14.70; MS(ESI)m/z: calcd for C9H10N3O2{[M+H]+}192.0768, found 192.0761。
4:白色固体,收率32%, m.p.165~167 ℃(文献[14]报道熔点为164~166 ℃);1H NMR(CDCl3, 300 MHz)δ: 9.51(dd,J=6.90 Hz, 1.80 Hz, 1H), 8.67(dd,J=4.20 Hz, 2.10 Hz, 1H), 8.39(s, 1H), 7.06(dd,J=6.90 Hz, 4.20 Hz, 1H), 4.37(q,J=7.20 Hz, 2H), 1.36(t,J=7.20 Hz, 3H);13C NMR(DMSO-d6, 75 MHz)δ: 160.00, 153.47, 150.89, 142.39, 136.46, 114.48, 111.67, 60.99, 14.73; MS(ESI)m/z: calcd for C9H10N3O2{[M+H]+}192.0768, found 192.0764。
(2)5的合成[15]
在反应瓶中加入3或4或两者混合物1.00 g(5.23 mmol)和乙醇(40 mL),搅拌使其溶解,加入水合肼0.28 mL(5.75 mmol),加热回流反应16 h。停止反应后,减压浓缩得到黄色固体,再用乙醚洗涤得到淡黄色固体5,收率100%, m.p.129~131 ℃(文献[8]报道熔点为130~133 ℃);1H NMR(DMSO-d6, 300 MHz)δ: 11.01(s, 1H), 7.20(s, 1H), 5.63(s, 2H), 4.16(q,J=7.20 Hz, 2H), 1.24(t,J=7.20 Hz, 3H);13C NMR(DMSO-d6, 75 MHz)δ: 161.03, 152.96, 130.31, 120.83, 59.53, 14.85; MS(ESI)m/z: calcd for C6H10N3O2{[M+H]+}156.0768, found 156.0761。
(3)6的合成[16]
在反应瓶中加入亚硝酸钠5.64 g(81.60 mmol)和水(18 mL),搅拌使其溶解,在-8 ℃时缓慢滴加溶有2.00 g(12.80 mmol)5的乙酸溶液(30 mL),滴加完毕保持-8 ℃反应12 h。停止反应后,用二氯甲烷(3×50 mL)萃取,有机相用无水硫酸钠干燥后减压浓缩得到粗品,经硅胶柱层析(洗脱剂:石油醚 ∶乙酸乙酯=2 ∶1,V∶V)纯化得到白色固体6,收率48%, m.p.136~138 ℃(文献[11]报道熔点为136~138 ℃);1H NMR(CDCl3, 300 MHz)δ: 7.69(s, 1H), 4.28(q,J=7.20 Hz, 2H), 1.27(t,J=7.20 Hz, 3H);13C NMR(CDCl3, 75 MHz)δ: 161.53, 133.31, 130.65, 127.05, 61.00, 14.31; MS(ESI)m/z: calcd for C6H8N3O4{[M+H]+}186.1465, found 186.1474。
(4)7和8的合成
在反应瓶中依次加入6(5.40 mmol),碘代试剂(6.48 mmol),碳酸钾(8.10 mmol)和乙腈(20 mL),混合后在65 ℃加热搅拌下反应15 h;停止反应后,冷却过滤,滤液浓缩后残留物经硅胶柱层析分离纯化得到7a~7d和8a~8d。
1-甲基-2-硝基-1H-咪唑-5-甲酸乙酯(7a):黄色固体,收率51%, m.p.55~57 ℃(文献[6]报道熔点为56~58 ℃);1H NMR(CDCl3, 300 MHz)δ: 7.52(s, 1H), 4.27(q,J=7.20 Hz, 2H), 3.61(s, 3H), 1.29(t,J=7.20 Hz, 3H);13C NMR(DMSO-d6, 75 MHz)δ: 161.74, 133.18, 130.95, 129.91, 60.28, 34.07, 14.71; MS(ESI)m/z: calcd for C7H9N3O4Na{[M+Na]+}222.0485, found 222.0491。
1-乙基-2-硝基-1H-咪唑-5-甲酸乙酯(7b):淡黄色黏稠液体,收率61%;1H NMR(CDCl3, 300 MHz)δ: 7.55(s, 1H), 4.29(q,J=7.20 Hz, 2H), 3.95(q,J=7.20 Hz, 2H), 1.38(t,J=7.20 Hz, 3H), 1.30(t,J=7.20 Hz, 3H);13C NMR(CDCl3, 75 MHz)δ: 162.02, 133.04, 132.46, 125.80, 60.77, 42.33, 15.31, 14.38; MS(ESI)m/z: calcd for C8H11N3O4Na{[M+Na]+}236.0642, found 236.0651。
1-丙基-2-硝基-1H-咪唑-5-甲酸乙酯(7c):黄色黏稠液体,收率62%;1H NMR(CDCl3, 300 MHz)δ: 7.55(s, 1H), 4.26(q,J=7.20 Hz, 2H), 3.87(t,J=7.20 Hz, 2H), 1.80~1.68(m, 2H), 1.28(t,J=7.20 Hz, 3H), 0.87(t,J=7.20 Hz, 3H);13C NMR(CDCl3, 75 MHz)δ: 162.04, 133.35, 132.33, 126.48, 60.78, 48.87, 23.37, 14.38, 10.85; MS(ESI)m/z: calcd for C9H13N3O4Na{[M+Na]+}250.0798, found 228.0786。
1-异丙基-2-硝基-1H-咪唑-5-甲酸乙酯(7d):黄色黏稠液体[17],收率80%;1H NMR(CDCl3, 400 MHz)δ: 7.61(s, 1H), 4.51~4.41(m, 1H), 4.30(q,J=7.20 Hz, 2H), 1.40(d,J=6.40 Hz, 6H), 1.31(t,J=7.20 Hz, 3H);13C NMR(CDCl3, 100 MHz)δ: 162.09, 132.57, 132.42, 122.87, 60.75, 49.38, 22.78, 14.39; MS(ESI)m/z: calcd for C9H14N3O4{[M+H]+}228.0979, found 228.0966。
1-甲基-2-硝基-1H-咪唑-4-甲酸乙酯(8a):淡黄色黏稠液体,收率26%;1H NMR(CDCl3, 300 MHz)δ: 7.53(s, 1H), 4.23(q,J=7.20 Hz, 2H), 3.81(s, 3H), 1.28(t,J=7.20 Hz, 3H);13C NMR(CDCl3, 75 MHz)δ: 159.67, 138.03, 136.10, 124.45, 60.73, 33.05, 14.29; MS(ESI)m/z: calcd for C7H9N3O4Na{[M+Na]+}222.0485, found 222.0494。
1-乙基-2-硝基-1H-咪唑-4-甲酸乙酯(8b):淡黄色黏稠液体,收率24%;1H NMR(CDCl3, 300 MHz)δ: 7.56(s, 1H), 4.33(q,J=7.20 Hz, 2H), 4.25(q,J=7.20 Hz, 2H), 1.30(t,J=7.20 Hz, 3H), 1.29(t,J=7.20 Hz, 3H);13C NMR(CDCl3, 75 MHz)δ: 159.41, 137.23, 136.46, 123.58, 60.69, 41.30, 15.33, 14.26; MS(ESI)m/z: calcd for C8H12N3O4{[M+H]+}214.0822, found 214.0828。
1-丙基-2-硝基-1H-咪唑-4-甲酸乙酯(8c):黄色黏稠液体,收率21%;1H NMR(CDCl3, 300 MHz)δ: 7.55(s, 1H), 4.27~4.20(m, 4H), 1.75~1.63(m, 2H), 1.28(t,J=7.20 Hz, 3H), 0.86(t,J=7.50 Hz, 3H);13C NMR(CDCl3, 75 MHz)δ: 159.19, 137.36, 136.24, 123.69, 60.46, 47.28, 23.37, 14.10, 10.67; MS(ESI)m/z: calcd for C9H14N3O4{[M+H]+}228.0979, found 228.0985。
1-异丙基-2-硝基-1H-咪唑-4-甲酸乙酯(8d):黄色黏稠液体,收率12%;1H NMR(CDCl3, 400 MHz)δ: 7.56(s, 1H), 5.40~5.30(m, 1H), 4.22(q,J=7.20 Hz, 2H), 1.51(d,J=7.20 Hz, 6H), 1.28(t,J=7.20 Hz, 3H);13C NMR(CDCl3, 100 MHz)δ: 158.52, 136.31, 135.80, 122.88, 59.67, 48.63, 19.59, 13.23; MS(ESI)m/z: calcd for C9H14N3O4{[M+H]+}228.0979, found 228.0987。
2 结果与讨论
2.1 合成路线分析
首先,以2-氨基嘧啶和3-溴丙酮酸乙酯为原料,发生分子间环化反应得到同分异构体3和4;接着,3和4无需分离,在水合肼作用下均生成5,这是由于咪唑环内双键会发生迁移生成更稳定的结构;然后,用亚硝酸钠和醋酸在低温下将氨基氧化至硝基生成6;最后,利用碘代烷烃,在碳酸钾作用下,即可实现一系列不同N-烷基取代衍生物的合成,但此时由于6咪唑环内双键同样会发生迁移而生成同分异构体7和8(图3)。
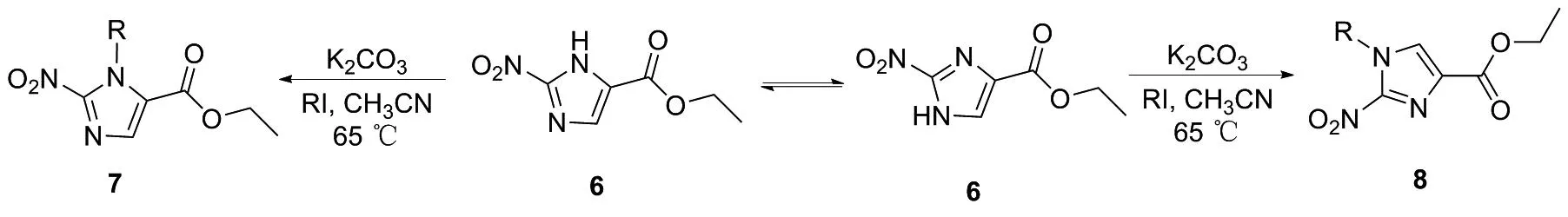
图3 同分异构体7和8的合成机制Figure 3 Mechanism of synthesis of isomers 7 and 8
2.2 取代基对异构体7和8的影响
为进一步考察不同取代基团对异构体7和8的影响,本文设计合成了甲基、乙基、丙基和异丙基4种不同空间位阻和电子效应的烷基取代产物,实验数据如表1所示。结果表明,随着取代基团空间位阻效应增大,空间位阻作用相对更大的7反而比8更有利于分子结构的稳定,说明该类结构稳定性的决定因素是电子效应而非位阻效应,即7比8具有更强的电子效应稳定性,并且随着取代基团的给电子能力增强,更加有利于7的生成。

表1 取代基对7和8合成比例的影响Table 1 Effect of substituents on the synthesis ratio of 7 and 8
本文以2-氨基嘧啶和3-溴丙酮酸乙酯为原料,设计合成了一系列1-烷基-2-硝基-1H-咪唑-5-甲酸乙酯衍生物(7a~7d)及其同分异构体1-烷基-2-硝基-1H-咪唑-4-甲酸乙酯衍生物(8a~8d)。该方法克服了传统合成路线中氮原子上取代基仅为甲基的局限性;通过对不同取代基团与分子结构稳定性的构效关系进行研究,结果表明:7比8具有更强的电子效应稳定性,从而为后续相关药物分子的结构修饰奠定了坚实基础。
