亚洲棉短纤维发育相关长链非编码RNA的鉴定及表达
2023-12-28王晓阳彭振邢爱双赵盈睿马欣丽刘方杜雄明何守朴
王晓阳,彭振,2,邢爱双,赵盈睿,马欣丽,刘方,2,杜雄明,2,何守朴,2
亚洲棉短纤维发育相关长链非编码RNA的鉴定及表达
王晓阳1,彭振1,2,邢爱双1,赵盈睿1,马欣丽1,刘方1,2,杜雄明1,2,何守朴1,2
1中国农业科学院棉花研究所/棉花生物育种与综合利用全国重点实验室,河南安阳 455000;2郑州大学农学院/棉花生物育种与综合利用全国重点实验室郑州基地,郑州 450001
【目的】长链非编码RNA(long non-coding RNAs,lncRNAs)是一类无蛋白质编码能力,但参与许多重要生命活动调控过程的长度大于200 nt的RNA。通过对亚洲棉无短纤维突变体(GA0149)和野生型(GA0146)纤维发育早期的转录组数据进行分析,挖掘调控短纤维发育的lncRNA,并明确其调控网络,为进一步解析棉花纤维发育机制奠定基础。【方法】选择GA0146和GA0149 2个材料在开花后当天(0 DPA)及花后3 d(3 DPA)、5 d(5 DPA)和8 d(8 DPA)的胚珠和纤维为材料进行转录组测序。鉴定lncRNA并预测其调控的靶基因;通过mRNA和lncRNA的差异表达分析,比较2个材料在不同纤维发育时期的差异。进一步利用KOBAS软件预测对差异lncRNA的靶基因进行富集分析并预测其参与的生物过程;最后通过实时荧光定量(RT-qPCR)技术对25个差异表达的lncRNA转录组数据进行验证。【结果】共鉴定获得15 339个lncRNA,其中11 595个lncRNA位于基因间区,包括2 428个反义lncRNA、350个内含子lncRNA及966个正义lncRNA。共有1 932个差异表达lncRNA(DE-lncRNA),它们所对应的8 134个靶基因中,有788个为差异表达基因(DE-mRNA)。KEGG代谢通路富集分析表明,DE-mRNA主要参与植物激素信号转导(plant hormone signal transduction)和内质网中蛋白质加工过程(protein processing in endoplasmic reticulum)。共表达调控网络分析显示,表达量差异比较显著的lncRNA()和其所调控的靶基因表达趋势一致,仅在野生型(GA0146)短纤维发育早期胚珠中特异表达;而lncRNA()与其调控的靶基因表达趋势相反,在突变体(GA0149)中的表达量显著高于野生型。RT-qPCR结果证实了转录组数据的真实性。【结论】鉴定了26个与亚洲棉短纤维发育相关的lncRNA,其通过调控植物激素信号转导途径相关的吲哚乙酸合成酶基因()和生长素响应蛋白基因()的表达而影响短纤维的发育。
亚洲棉;短纤维突变体;长链非编码RNA;调控网络;荧光定量PCR
0 引言
【研究意义】棉花纤维起始于种子表皮的一个单细胞,是纺织工业的主要原料。棉花纤维的产量主要取决于起始阶段,该时期决定了胚珠纤维的数量。尽管所有棉花胚珠表皮细胞都具有分化成纤维的潜在能力,但最终只有30%的细胞能够分化成纤维[1]。因此,可通过调节纤维起始过程表皮细胞数量来提高纤维产量。根据棉花成熟纤维长度可以分为长纤维(Lint)和短纤维(Fuzz),具有重要经济作用的长纤维起始于开花当天(day post anthesis,0 DPA),最终长度能达到2.5—3.5 cm,短纤维起始于开花后3—5 DPA,长度为5—10 mm[2]。虽然短纤维的起始要晚于长纤维的起始,但是长纤维和短纤维在分化过程可能经历相似的分子机制。因此,研究亚洲棉短纤维起始及其相关lncRNA的表达调控对解析棉花纤维发育具有重要的理论意义和实践指导作用。【前人研究进展】天然纤维突变体由于其特殊的性状,是解析长纤维和短绒发育机制的重要种质资源[3-7]。目前,科学家对四倍体陆地棉纤维突变体进行了大量研究,而对二倍体亚洲棉突变体的研究较少。与四倍体棉花相比,二倍体亚洲棉基因组结构较为简单,因此,适合用于挖掘光籽性状关键基因及阐明短绒发育的分子机制[8-10]。前期杜雄明课题组利用215份亚洲棉重测序数据进行短纤维性状的全基因组关联分析以及转基因试验,确定()可能是控制亚洲棉短纤维发育的候选基因[11]。随后,Feng等[12-13]利用QTL定位和转录组分析方法也鉴定出()及编码Fasciclin-like阿拉伯半乳聚糖蛋白18(FLA18)和转运蛋白的基因为控制短纤维性状的候选基因。尽管已经鉴定出许多参与纤维发育过程的基因,但是,控制纤维起始的整个调控网络目前还未解析。长链非编码RNA(long noncoding RNAs,lncRNAs)是一类长度大于200 bp,不具备蛋白编码能力的RNA分子,与mRNA类似,都是由RNA聚合酶Ⅱ(RNA pol Ⅱ)进行反转录,在结构上具有5′加帽、3′聚腺苷酸化等特点[14-15]。与mRNA不同的是,lncRNA通常是从基因组的基因间区域转录的,而一些lncRNA来源于编码基因的反义链[16]。lncRNA在不同植物的基因表达和维持染色体稳定性方面起重要调控作用,参与广泛的生物调节过程,例如拟南芥开花过程、水稻光敏雄性不育、沙棘果维生素C合成、光诱导的苹果种皮花青素合成,以及植物相应逆境和非逆境生物胁迫等[17-26]。随着多个棉花品种的高质量基因组数据的发布[27-28],前人已运用RNA-seq和生物信息分析方法鉴定出一批棉花lncRNA。Wang等[29]和闫飞林[30]通过对海岛棉中lncRNA进行系统分析,鉴定出30 550个基因间lncRNA、4 718个反义lncRNA和35 268个反义lncRNA(lncNATs),经组织特异性分析,发现约76%的同源1ncRNA表现出At或Dt亚组的偏好性表达模式;通过调查转基因RNAi株系表型,发现lncRNA棉花转基因株系的纤维均有变短趋势,说明lncRNA可能负调控棉纤维的起始和伸长。通过对亚洲棉纤维起始期和快速伸长期的转录组数据进行分析,发现51.9%的基因间lncRNA(lincRNAs)和54.5%的lncNAT倾向于在纤维发育的一个特定阶段优先表达[31]。通过分析Xuzhou142及其纤维突变体,以及由这两个材料杂交产生的3个不同衣分产量株系0 DPA和5 DPA的转录组数据,鉴定出35 802个lncRNA,其中645个lncRNAs在中优先表达,病毒诱导的基因沉默试验证实XLOC_545639和XLOC_039050能增加胚珠细胞起始数目[32]。通过对长纤维突变体()和野生型开花0 DPA和8 DPA的转录组数据进行分析,鉴定出18 333个lncRNA,差异分析和GO富集通路分析发现,差异lncRNA的靶基因主要参与脂肪酸生物合成和延伸途径[33]。同时,一些参与陆地棉盐胁迫过程的lncRNA也被鉴定,拟南芥转基因和病毒诱导的基因沉默试验证明,lncRNA973能提高陆地棉的耐盐性[34]。目前,利用多策略RNA-seq技术对亚洲棉中的lncRNA进行全面地功能注释和鉴定,并对其转录调控机制进行了解析,为理解棉花lncRNA的功能提供了宝贵研究资源[35]。【本研究切入点】目前,尽管关于棉花短纤维候选基因的挖掘已经取得了很大进展,但其发育机制和调控网络尚不清楚。lncRNA在调控植物生长发育中起不可或缺的作用,但对其调控棉花纤维起始和伸长的机制还知之甚少。【拟解决的关键问题】本研究通过分析亚洲棉无短纤维突变体(GA0149)和其野生型(GA0146)纤维发育早期的转录组数据,鉴定差异表达lncRNA,明确其功能性特征及表达水平,为进一步研究lncRNA在棉花纤维发育中的作用提供依据。
1 材料与方法
1.1 试验材料
亚洲棉野生型中美棉GA0146既有长纤维又有短纤维,其近等基因系短纤维突变体GA0149有长纤维没有短纤维,二者来自于中国农业科学院棉花研究所国家棉花种质资源中期库,经过大约连续10代自交,均没有出现短纤维性状的分离,其他农艺性状接近。所有材料均在中国农业科学院棉花研究所安阳试验田种植,并按照标准的田间生产管理方法。开花前一天自交,同时挂牌记录开花时间,即为-1 DPA(开花前一天)。在上午分别取每个材料0 DPA(开花当天)、3 DPA、5 DPA和8 DPA的胚珠或纤维,液氮速冻,-80 ℃冷冻保存,用于RNA的提取和转录组测序。每个时期设置3个生物学重复。
1.2 RNA提取和转录组测序
使用多糖多酚植物总RNA提取试剂盒(No. DP441)提取GA0146和GA0149短纤维发育早期0、3和5 DPA的胚珠及8 DPA的胚珠和纤维混合物,经1.2%琼脂糖凝胶电泳检测完整性,用NanoDrop-2000(Thermo Scientific)分光度计测定浓度,然后用于RT-qPCR分析和cDNA文库构建。利用Illumina HiSeq2000平台进行双末端测序,测序读长为PE150,获得原始数据。
1.3 亚洲棉lncRNA的生物信息学预测和表达分析
将原始数据通过FastQC软件去掉接头和低质量的数据,获得1 064.24 Gb高质量clean reads(Q>20),各样品Clean Data均达到16.45 Gb,Q30碱基百分比在86.17%及以上。利用TopHat2软件[36]将clean reads与亚洲棉石系亚1号三代基因组[28]比对。利用HTSeq v0.6.1软件与外显子上的reads数进行比对、计数[37]。通过Cufflinks 2.0程序组装每个转录本。然后,利用Cuffdiff组装来自BAM输出文件的所有转录本,使用每千碱基转录物每百万映射读取的片段(fragments per kilobase of transcript per million fragments mapped,FPKM)值来确定转录本丰度[38]。采用嵌入到CUFFLINKS软件中的多次读数功能和片段偏差校正方法来提高基因表达水平估计的准确性。
根据起始位置,lncRNA可以分为基因内lncRNA(intronic long non-coding RNAs,intronic-lncRNAs)、反义lncRNA(antisense long noncoding RNAs,antisense- lncRNAs)、正义lncRNA(sense lncRNAs)和基因间lncRNA(intergenic long non-coding RNAs,lincRNAs)等4种类型[39]。通过以下6个步骤鉴定lncRNA的类型:(1)丢弃在少于2个样本中被检测到的同时用2种方法组装的转录本;(2)删除覆盖率小于转录本长度一半的转录本;(3)去除由rRNA和tRNA延伸而来的转录本(cutoff E-value<0.001);(4)去除长度小于200 bp的转录组;(5)利用CPC(Coding Potential Calculator)和CNCI(Coding-Non- Coding Index)软件排除具有编码能力的转录本[40];(6)将剩余的转录本在Swiss-Prot和Pfam数据库中进行比对,以消除具有蛋白编码能力和蛋白结构域的转录本。
1.4 靶标基因预测
为了研究lncRNA的功能,对其作用于相邻基因的靶基因进行预测。在lncRNA上下游10 kb内搜索每个lncRNA周围相邻的编码基因,研究它们的功能。通过基因表达水平来预测lncRNA和其靶基因的相互作用。使用R工具,应用皮尔逊相关系数来阐明lncRNA和mRNA之间的相对表达关系,选择皮尔逊相关系数(2)≥0.95。然后,利用lncRNA和mRNA的相关参数构建共表达网络,揭示lncRNA和mRNA之间的相互作用,并用Cytoscape软件(3.8.2)将共表达网络可视化;利用goseq和KOBAS软件对lncRNA的靶基因功能进行富集[41-42]。
1.5 差异表达基因(mRNA)和lncRNA分析
运用“Deseq2”R软件包对野生型和突变体同一发育时期的mRNA和lncRNA的FPKM和count值进行差异表达分析,同时,过滤掉FPKM<0.5的lncRNA[43],筛选差异表达基因(differentially expressed mRNA,DE-mRNA)和lncRNA(DE-lncRNA)。将|log2(fold change)|>1和-value<0.05作为筛选DE-lncRNA的标准。
1.6 KEGG功能富集分析
为了解析差异基因的功能,将获得的DE-lncRNA的靶基因在数据库中进行KEGG(kyoto encyclopedia of genes and genomes)富集通路分析。使用KOBAS软件检验KEGG途径中DE基因或lncRNA靶基因的富集因子,选择-value<0.05的富集通路。
1.7 转录组数据的RT-qPCR分析
为了验证转录组数据的准确性,挑选25个在4个时期共有的差异lncRNA进行RT-qPCR验证。利用在线软件NCBI-Primer(http://www.ncbi.nlm.nih.gov/ tools/primer-blast/)进行lncRNA qRT-PCR引物设计(电子附表1)。使用EvoM-MLV预混型(AG11728)反转录试剂盒(含去除gDNA试剂,用于qPCR)将1 μg总RNA合成cDNA,以为内参,进行实时荧光定量PCR验证。每个基因至少设置3个技术重复,3个生物学重复。以GA0146材料0 DPA样品的基因表达量为1,用2-ΔΔCT方法[44]分析突变体和野生型各个时期的lncRNA相对表达量。
2 结果
2.1 亚洲棉短纤维突变体(GA0149)和其野生型(GA0146)中的lncRNA鉴定
前期通过观察亚洲棉短纤维起始早期的表型观察,在3 DPA时突变体胚珠表皮未发现细胞突起,而野生型有大量细胞突起,说明短纤维可能起始于3 DPA[45]。当胚珠长至成熟期,对其种子表型进行调查,发现野生型GA0146成熟种子表皮同时附着短纤维和长纤维,而其短纤维突变体GA0149成熟种子表皮则仅附着长纤维(图1-A)。为了进一步了解造成种子短纤维表型差异的原因,选取亚洲棉野生型和其突变体短纤维发育关键时期(0 DPA、3 DPA、5 DPA和8 DPA)的胚珠及纤维通过Illumina平台进行全转录组测序,鉴定调控短纤维发育的关键lncRNA。将获得数据与亚洲棉参考基因组比对,共获得15 339个lncRNA。根据与编码基因的相对位置,鉴定出11 595个基因间lncRNA、2 428个反义lncRNA、350个基因内lncRNA和966个正义lncRNA(图2-B)。
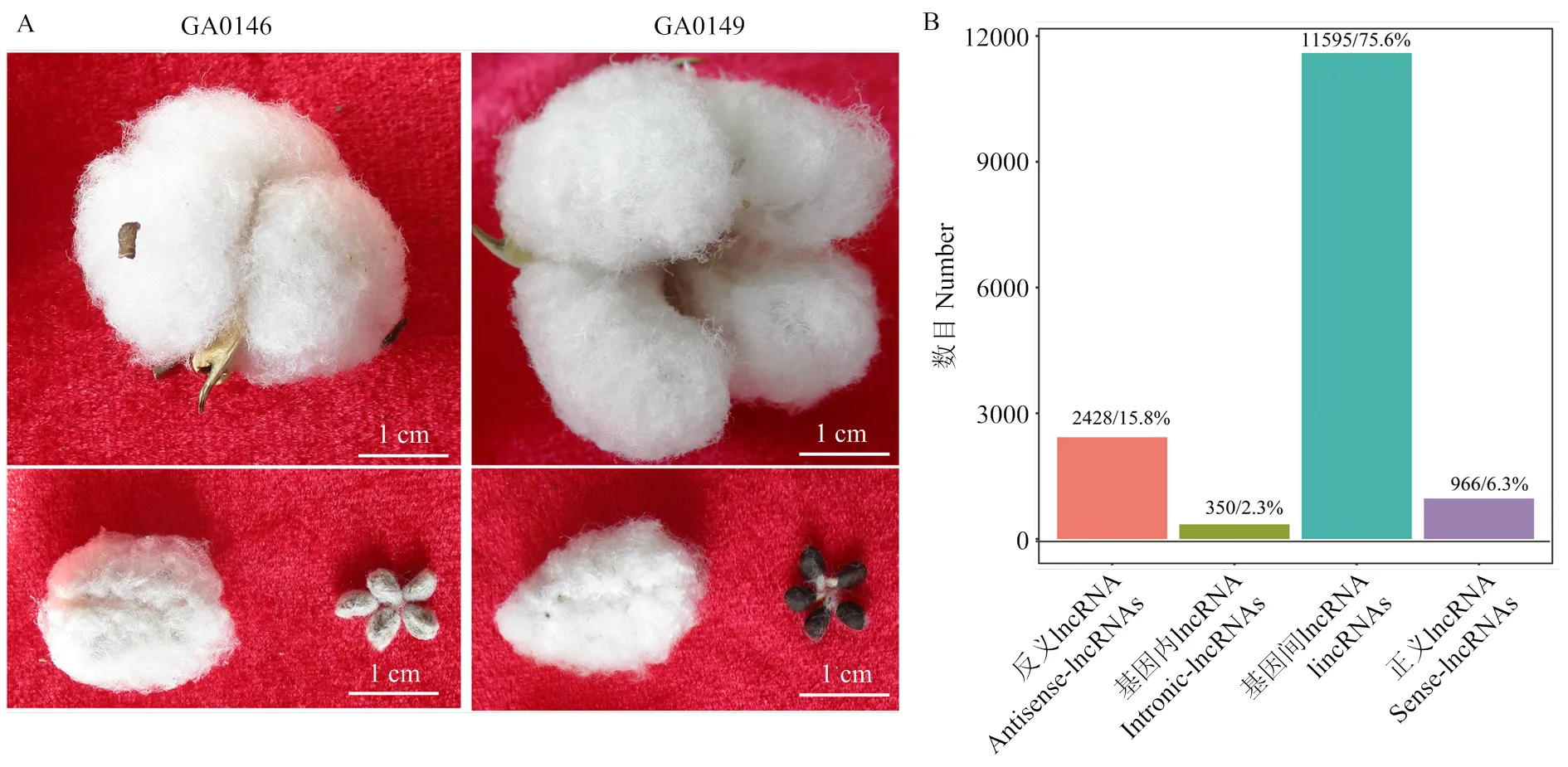
A:种子成熟期的亚洲棉野生型和短纤维突变体表型;B:4种类型lncRNA的分布
GC含量与四倍体棉花基因组中亚基因组间的基因偏向性表达有显著相关性[29]。通过对lncRNA和mRNA的GC含量、转录本长度以及外显子数目进行分析,与mRNA相比,lncRNA的GC含量更低(图2-A);同时,发现mRNA的平均外显子长度要大于lncRNA的外显子长度,lncRNA外显子变化范围为200—11 619 bp,mRNA的变化范围为200—25 929 bp(图2-B);大部分lncRNAs含有2个外显子,而mRNA含有多个外显子(图2-C)。lncRNA对mRNA的调控主要分为顺式和反式调节,通过分析调控方式发现,大部分lncRNA对mRNA起顺式调控作用,少部分为反式调节或者为2种方式的调节(图2-D)。

A:lncRNA和mRNA中的GC含量;B:lncRNA和mRNA中的外显子长度分布情况;C:每个lncRNA和mRNA转录本的外显子数目;D:lncRNA和mRNA之间的相互作用方式
2.2 亚洲棉短纤维起始期差异lncRNA的表达分析
为了鉴定出参与短纤维发育的候选lncRNA,对亚洲棉野生型和其突变体4个纤维发育时期(GA0149 vs GA0146 0 DPA、3 DPA、5 DPA和8 DPA)的lncRNA转录本表达量进行分析,共鉴定出1 932个差异表达lncRNA,其中,612个来自于短纤维发育5 DPA,520个来自于长纤维发育0 DPA,570个来自于短纤维发育3 DPA,440个来自于短纤维发育8 DPA,短纤维发育3和5 DPA共有的DE-lncRNA数目为167个(图3和图4-A)。火山图(图3)显示,4个比较组中,DE-lncRNA的基因表达谱,上调表达和下调表达的lncRNA具有明确的分布模式,表明不同比较组中存在不同的表达模式。4个时期共有的DE-lncRNA数目为65个(图4-B、表1和电子附表2)。其中,有28个lncRNA在突变体中发生上调表达,37个lncRNA在突变体中发生了下调表达(表1)。说明3和5 DPA共有的DE-lncRNA可能调控短纤维的起始和发育;在短纤维起始和发育阶段,4个时期共有的65个DE-lncRNA的表达在2个材料中保持趋势一致,同时,可以证明这些共有的DE-lncRNA可能调控长纤维起始和发育。
2.3 与短纤维起始相关的差异lncRNA的功能分析
为了解析lncRNA的表达模式与短纤维发育的关系,分析野生型和突变体同一纤维发育时期的lncRNA表达情况(图5-A—B)。在长纤维起始期(0 DPA),突变体中有261个lncRNA发生了上调表达,259个lncRNA发生了下调表达,说明有大量lncRNA参与了长纤维的起始;在短纤维起始期(3 DPA),有316个lncRNA在突变体中发生了上调表达,254个lncRNA在突变体中发生了下调表达;在短纤维发育期(5 DPA),有300个lncRNA在突变体中发生了上调表达,312个lncRNA在突变体中发生了下调表达;在纤维伸长期(8 DPA),有179个lncRNA在突变体中发生了上调表达,261个lncRNA在突变体中发生了下调表达。说明在短纤维起始期发生上调表达或下调表达的差异lncRNA多于长纤维起始期和伸长期的差异lncRNA。
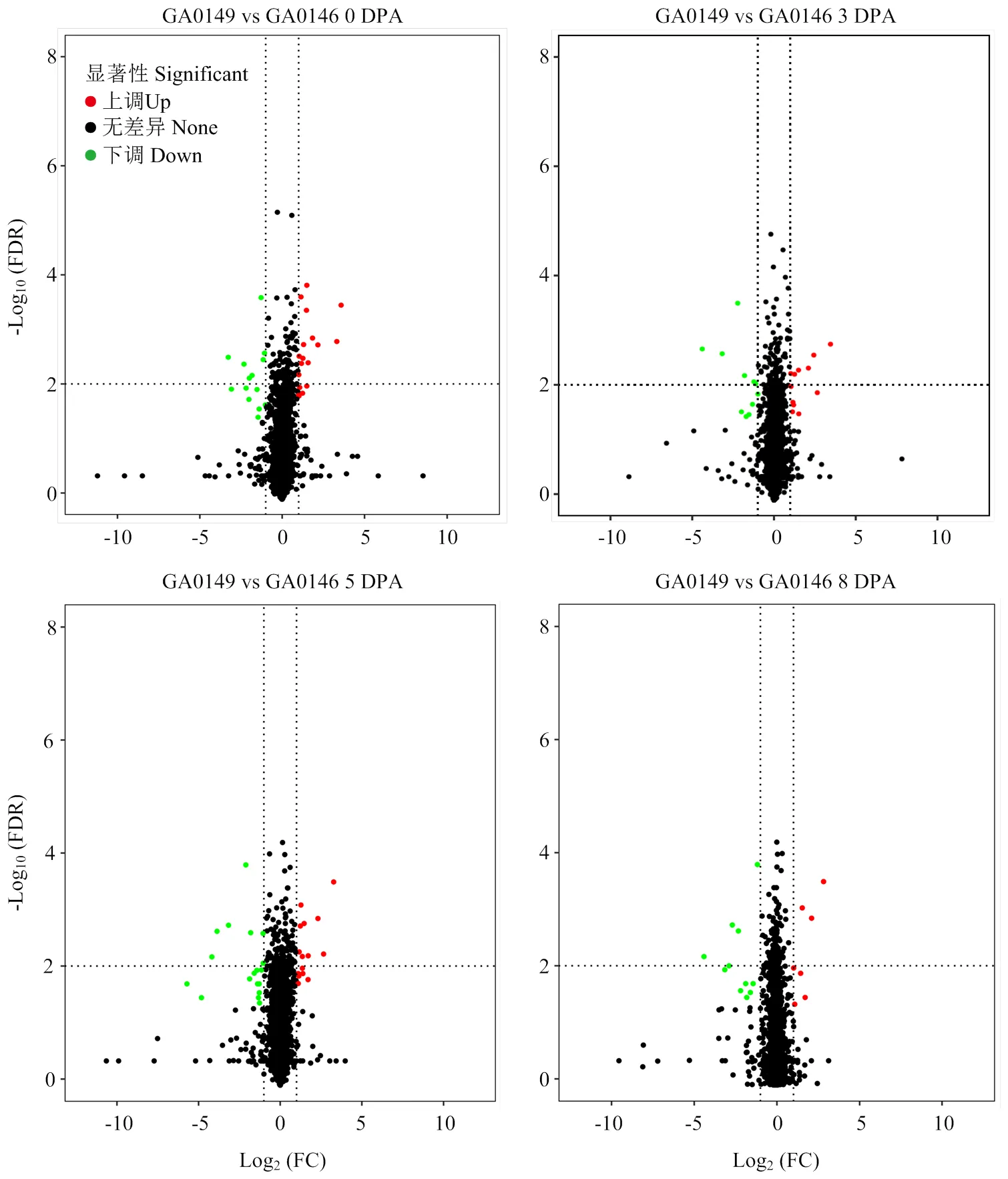
图3 火山图展示lncRNA表达量变化和FDR值之间的分布
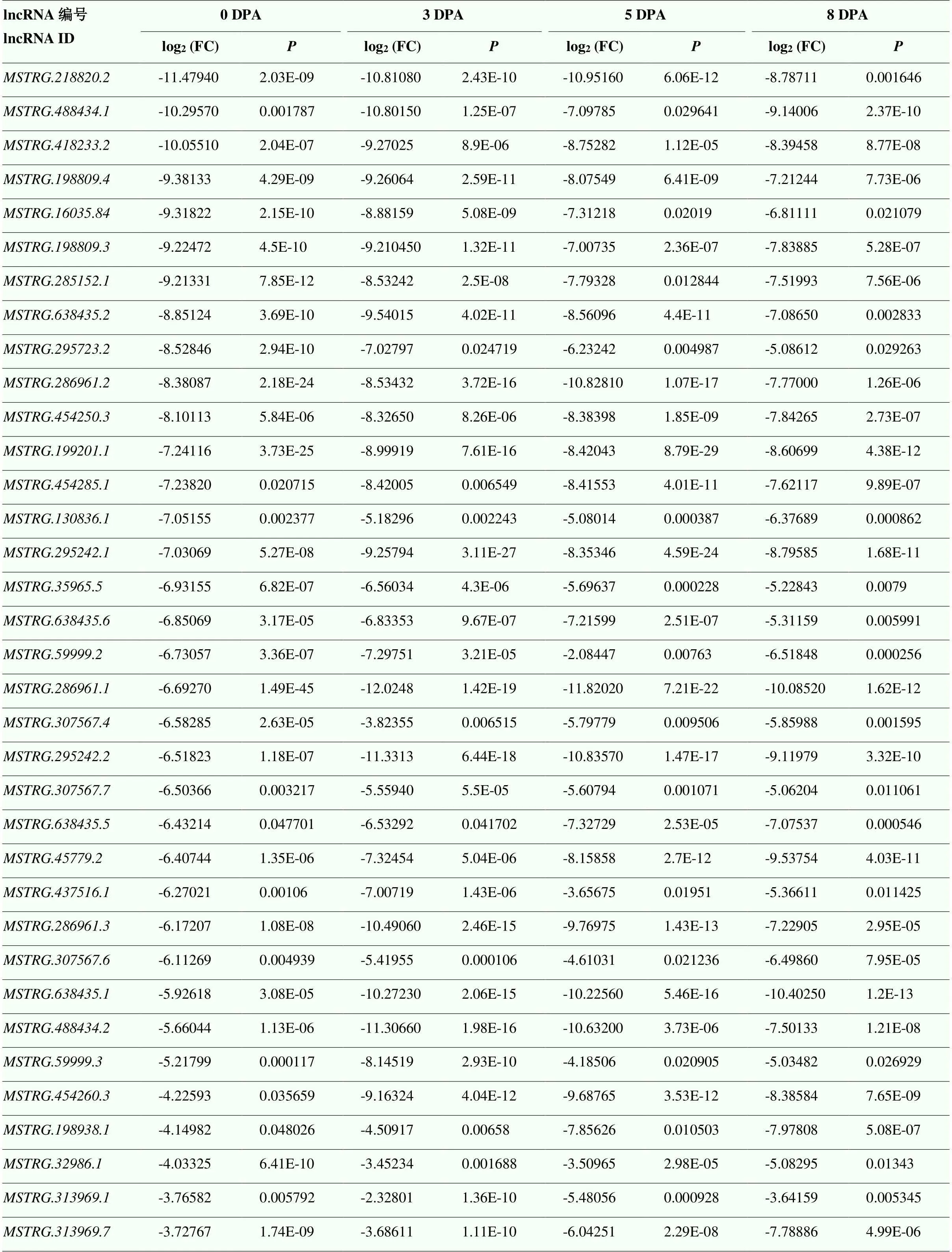
表1 亚洲棉短纤维突变体(GA0149)和其野生型(GA0146)短纤维发育早期共有差异lncRNA及其表达量变化倍数
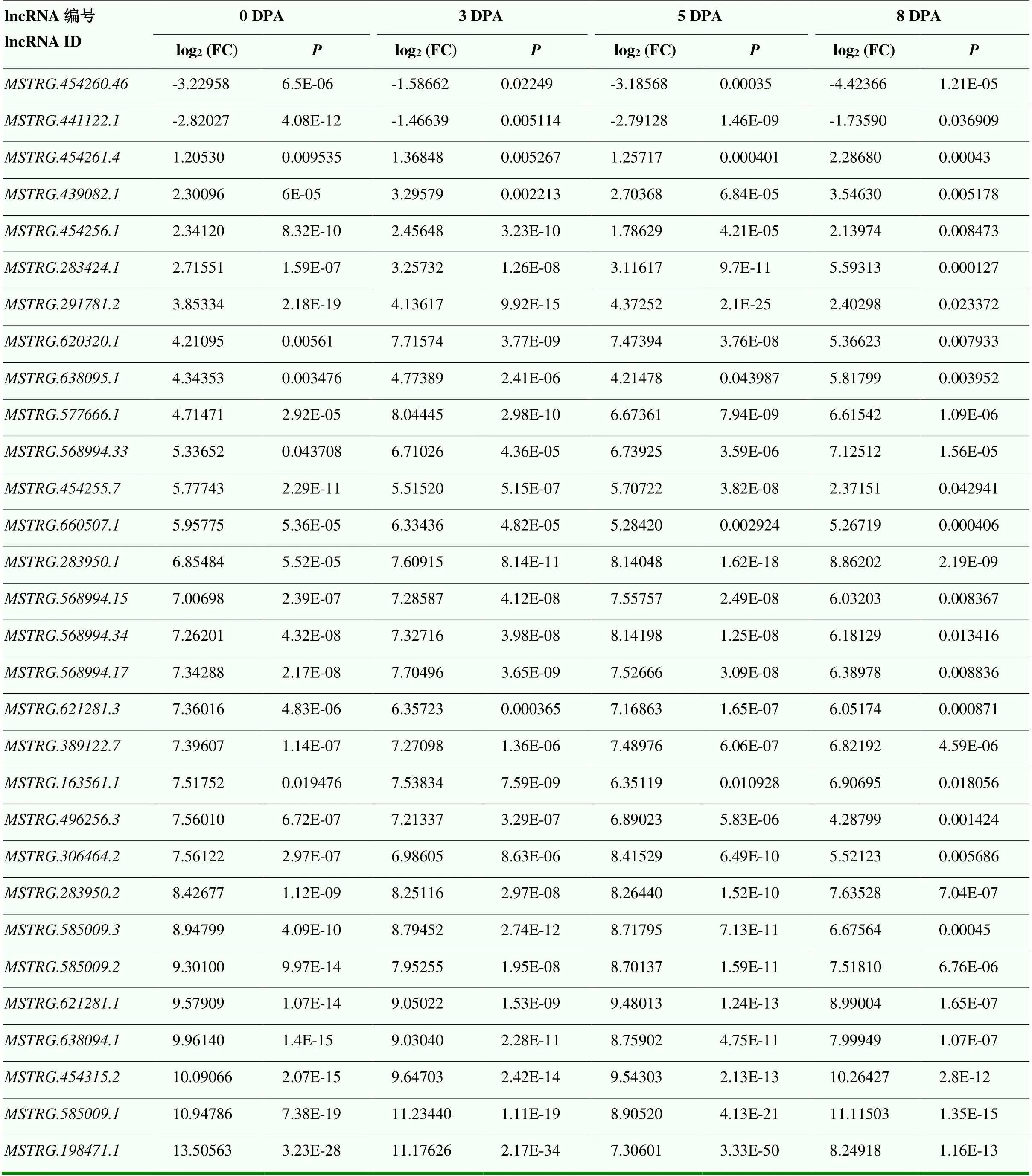
续表1 Continued table 1
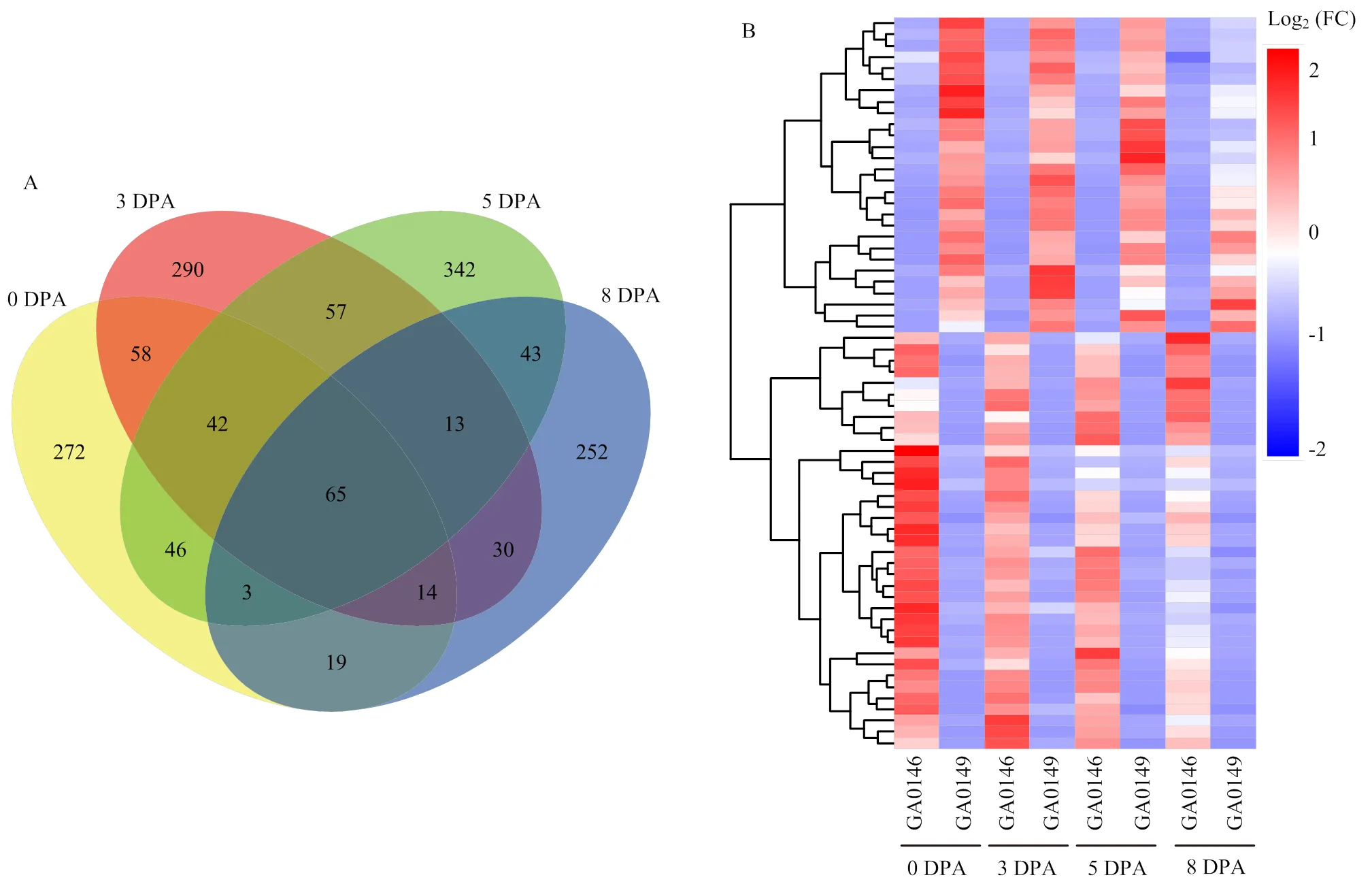
A:2个材料4个纤维发育时期共有差异lncRNA的韦恩图;B:65个共有的差异lncRNA表达量热图
为了解短纤维发育早期各时期lncRNA表达情况,比较2个材料相邻2个时期lncRNA的表达变化(图5-A—B)。在GA0146中,在长纤维起始期至短纤维起始期(0—3 DPA),291个lncRNA发生上调表达,201个下调表达;在短纤维发育初期(3—5 DPA),254个lncRNA上调表达,189个下调表达;在短纤维发育初期至长纤维伸长期(5—8 DPA),334个lncRNA上调表达,426个下调表达。而在短纤维突变体GA0149中,从0—3 DPA时期,344个lncRNA上调表达,199个下调表达;在3—5 DPA时期,271个lncRNA上调表达,284个下调表达;在5—8 DPA时期,435个lncRNA上调表达,805个下调表达。总之,在长纤维起始期至短纤维起始期,上调表达的lncRNA多于下调表达的lncRNA,说明大部分lncRNA可能正调控长纤维起始;在短纤维发育初期至长纤维伸长期,差异lncRNA较少;在纤维伸长期,差异lncRNA最多,并且下调表达的lncRNA多于上调表达的lncRNA,表明大部分lncRNA可能负调控纤维伸长,同时,由于伸长期的纤维细胞生长最快,并且该时期主要影响纤维长度、强度和细度等品质性状,因此,在纤维伸长期,lncRNA的表达水平可能发生了较大变化。
为了研究lncRNA的功能,根据在染色体上的位置,预测差异lncRNA所调控的靶基因。共筛选到8 134个mRNA,同时按照差异基因分析标准,对mRNA的表达量进行计算以及差异性分析,最终鉴定到788个差异表达的靶基因。经KEGG代谢通路富集显示(图5-C),差异靶基因主要参与的通路如:植物激素信号转导(plant hormone signal transduction)和内质网中蛋白质加工过程(protein processing in endoplasmic reticulum)。对富集到植物激素信号转导通路中的14个差异基因表达量进行分析(表2),发现8个差异基因参与生长素响应和生物合成过程,与野生型相比,生长素响应蛋白基因在突变体的开花当天上调表达,而在3、5和8 DPA表达水平没有发生变化;吲哚乙酸合成酶基因在突变体的5 DPA表达量上升,其他时期的表达无显著差异;生长素响应蛋白编码基因和,在突变体的5 DPA表达量上升,8 DPA表达量下降,其他时期表达量没有变化,表明生长素对短纤维的起始可能没有影响,对纤维的伸长起正调控作用。参与茉莉酸甲酯合成途径的转录因子MYC基因()在突变体5 DPA的表达量发生了显著上调表达,说明该转录因子可能负调控短纤维的起始。参与乙烯合成途径的基因以及参与脱落酸(ABA)合成途径的基因在突变体的5 DPA表达量上升,8 DPA表达量显著下降,说明过量的乙烯和ABA能抑制短纤维起始,少量的乙烯能促进纤维伸长。同时,也发现水杨酸(SA)和赤霉素(GA)合成途径的基因也参与了纤维发育过程。说明植物激素在纤维起始和伸长过程中起重要作用,这些可能的靶基因为lncRNA在棉花纤维发育中的作用提供了新见解。

A:短纤维发育早期时期上调表达和下调表达差异lncRNA数目,红色代表上调的lncRNA,蓝色代表下调的lncRNA;B:4个纤维发育时期差异lncRNA表达量热图;C:差异lncRNA的靶基因KEGG富集通路
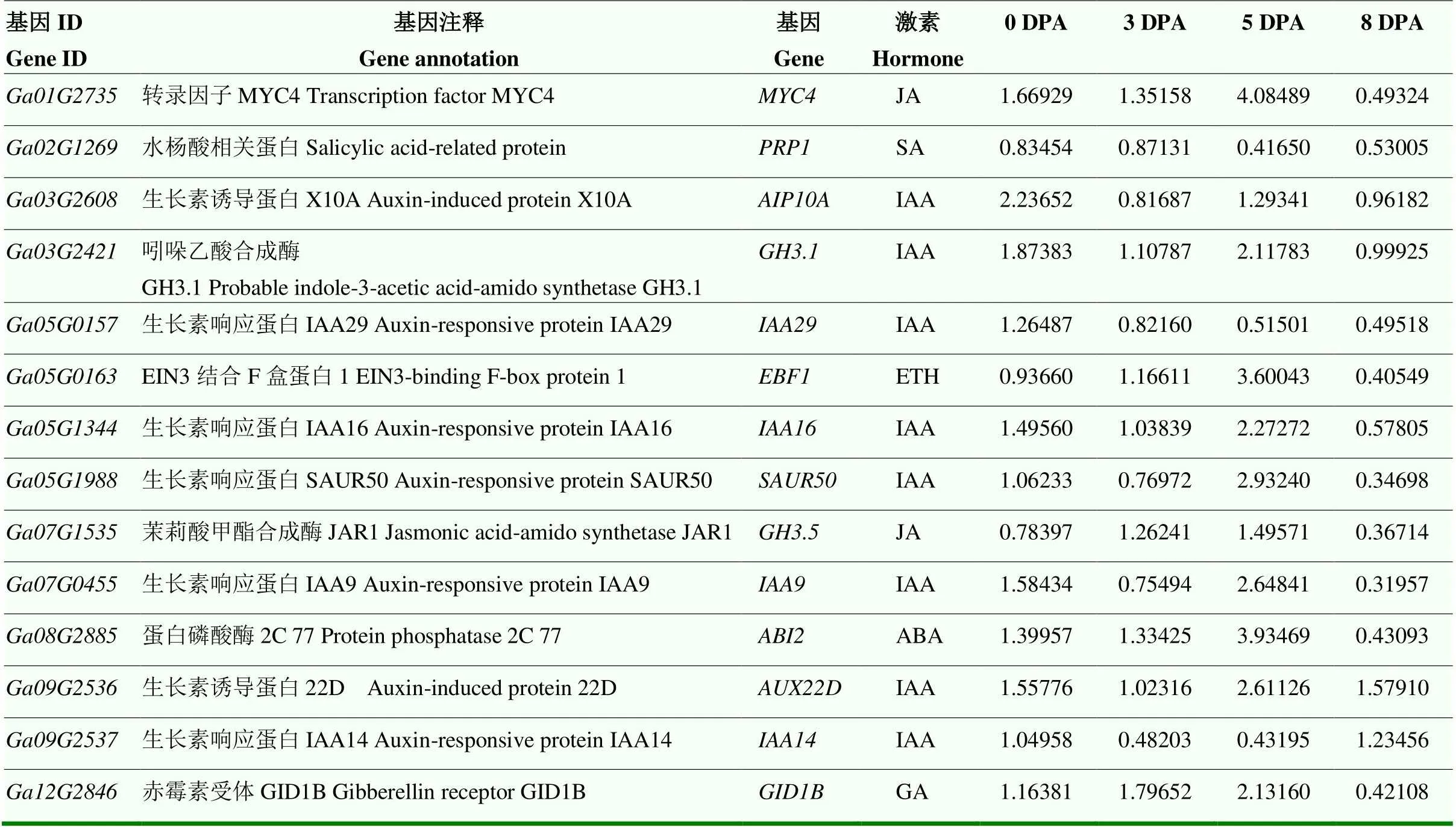
表2 植物激素信号通路的差异靶基因及其表达情况
2.4 lncRNA和其靶基因的调控网络分析
lncRNA的一个主要功能是顺式调控同一等位基因上的相邻基因,从而调节基因的转录或转录后表达。因为顺式作用的lncRNA靶向邻近基因,在所有已鉴定的lncRNA上、下游10 kb区域搜索了编码基因,同时预测它们的功能。通过分析具有顺式调控作用的lncRNA,构建lncRNA和mRNA的共表达网络。大多数mRNA和lncRNA是一对一的匹配。然而,大部分lncRNA也可以调控多个mRNA。考虑到表格无法显示lncRNA和mRNA之间的大量网络信息,选择了一些mRNA与lncRNA以绘制它们之间的网络图(图6)。结果显示,lncRNA()可以同时调控12个mRNA的表达,而则调控18个mRNA的表达;同时发现一个mRNA也可以受多个lncRNA的调控,如6个lncRNA调控编码基因的表达,则受22个lncRNA的调控。
位于蛋白质编码基因上游的lncRNA可能与启动子区域或者顺式调节元件有重叠,并可能在转录或转录后水平调节其附近基因的表达。位于蛋白质编码基因下游的lncRNA可以从基因的3′UTR或下游区域启动转录,并可能参与基因间相互调节作用。为了进一步解析差异lncRNA对其下游靶基因表达的影响,随机选择4个差异表达明显的lncRNA及其所对应的靶基因进行网络调控分析(图7)。lncRNA()仅在野生型短纤维发育早期表达,在短纤维突变体的各个时期都不表达,而其所调控的靶基因(功能未知蛋白)、(功能未知蛋白)和(含五肽重复序列的蛋白)也是在野生型中显示高表达,在突变体中几乎不表达;(功能未知蛋白)和(功能未知蛋白)则与lncRNA的表达趋势相反,在突变体中的表达水平高于其在野生型中的表达水平,说明MSTRG.454250.3通过正调控、和的表达,负调控和的表达,进而抑制纤维的发育。仅在野生型中表达的lncRNAs()与其下游靶基因的表达趋势一致,正调控下游靶基因的表达。正调控下游靶基因的表达,在野生型中的表达量高于突变体中的表达量。与上述3个lncRNA的表达趋势相反,在突变体中的表达量高于野生型中的表达量,同时正调控下游靶基因的表达,说明该lncRNA在短纤维起始和发育过程起负调控作用。
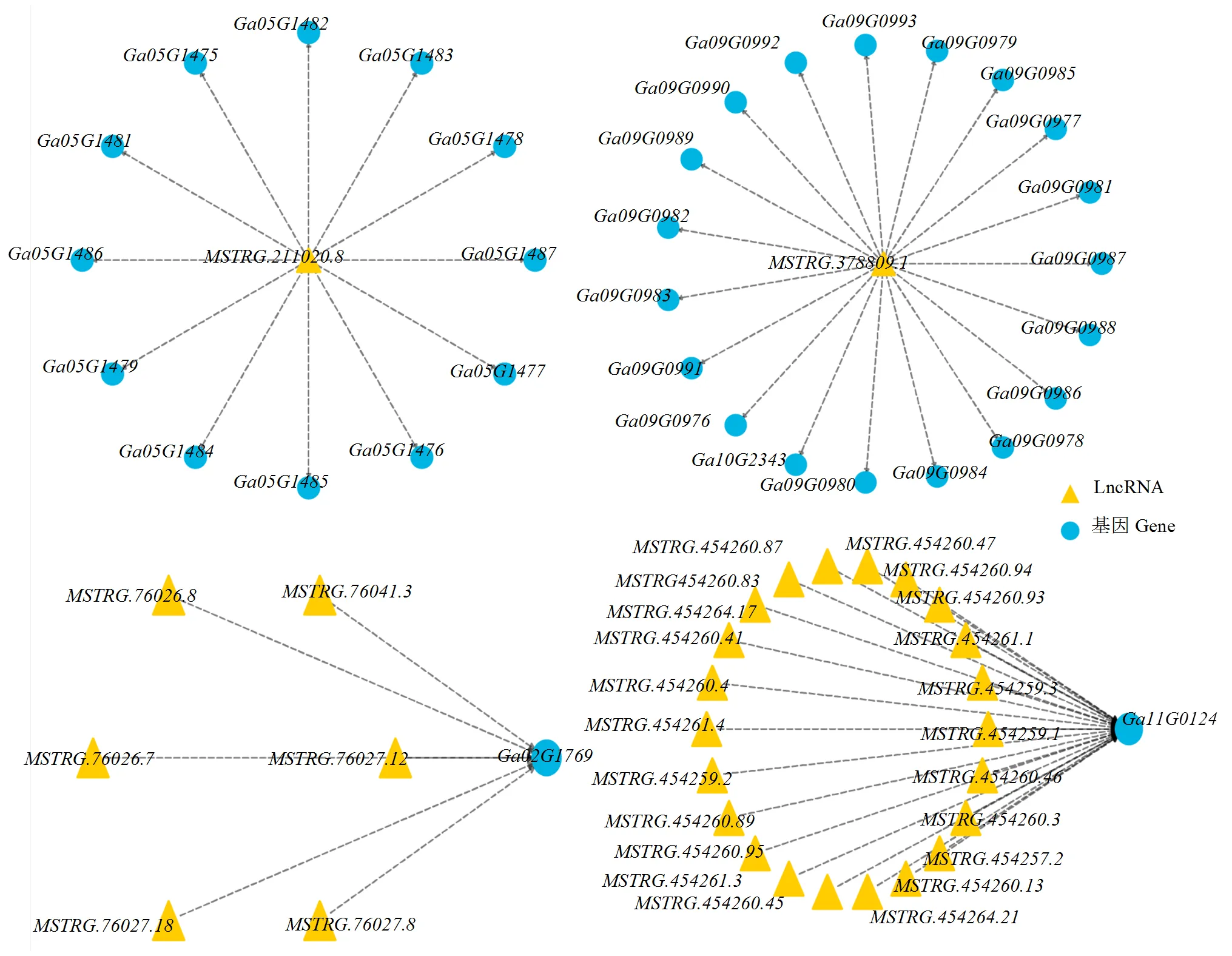
图6 lncRNA与mRNA之间的相互作用关系
2.5 参与短纤维起始的lncRNA表达量验证
长链非编码RNA在许多植物的生长、发育和适应机制中发挥着重要作用,如花发育、有性繁殖、果实成熟、纤维发育、生物胁迫和非生物胁迫反应。为了揭示lncRNA在亚洲棉短纤维发育中的作用,结合RNA-seq中的lncRNA表达水平,随机选择25个lncRNA在亚洲棉野生型(GA0146)和其短纤维突变体(GA0149)的0、3、5和8 DPA胚珠及纤维中进行荧光定量PCR验证。转录组数据与RT-qPCR结果一致(图8),一些lncRNA(如、、、和)倾向于在突变体4个纤维发育时期(GA0149)中高表达,在野生型(GA0146)中不表达,说明这些lncRNA可能在棉花短纤维发育中起负调控作用。另外一些lncRNA(如、、、和)在亚洲棉野生型(GA0146)的0、3、5和8 DPA时期高于其在突变体中的表达量,说明这些lncRNA正调控棉花短纤维的发育。
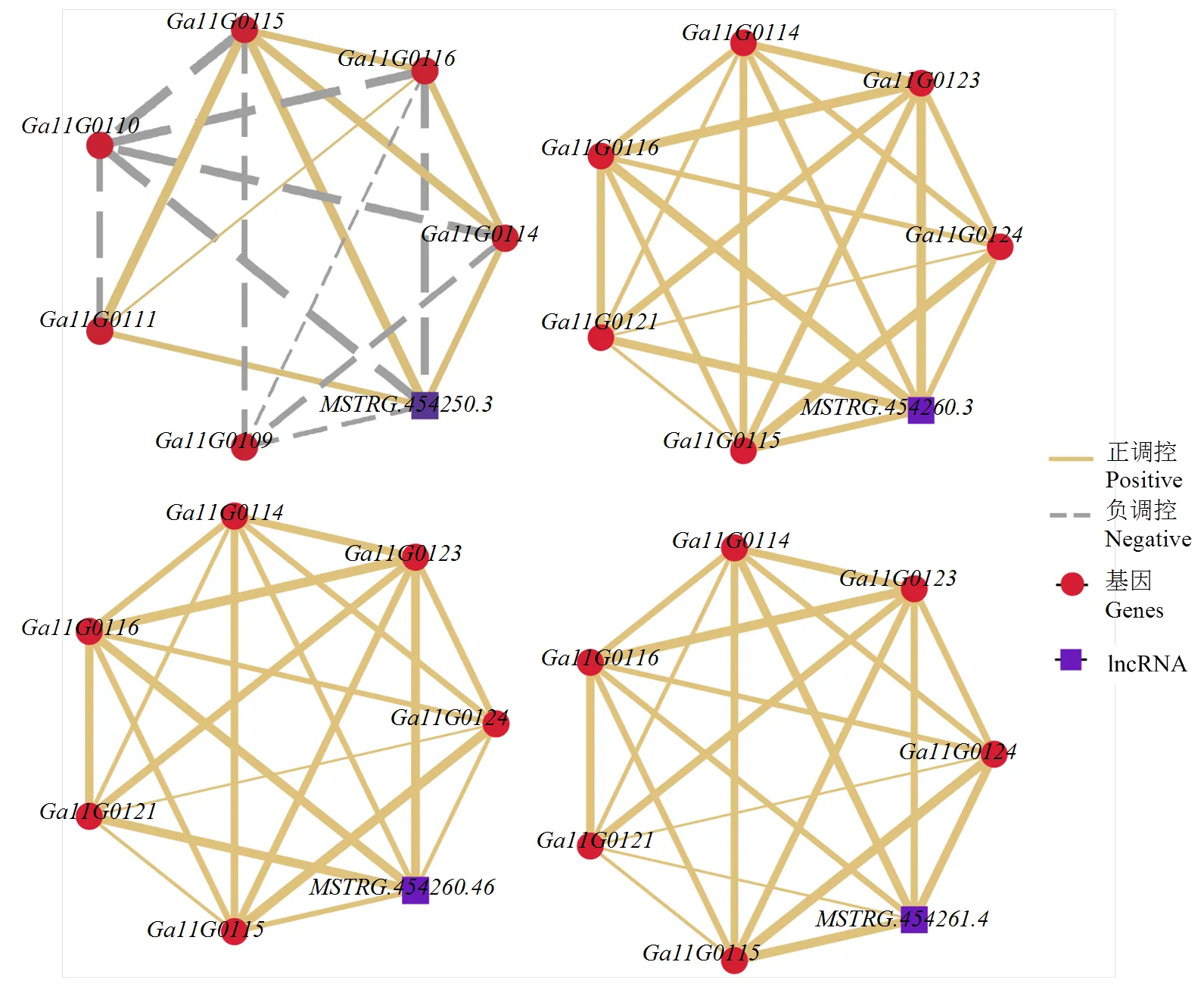
图7 亚洲棉野生型和短纤维突变体4个纤维发育时期的差异lncRNA和其差异靶基因的共调控网络
2.6 二倍体和四倍体棉花中lncRNA的鉴定
通过比较二倍体和四倍体棉种间lncRNA序列以及使用共线分析方法,在高置信水平上鉴定具有保守性低的同源lncRNA位点,找出亚洲棉和陆地棉中共有及特有的lncRNA,结果显示,在2个棉种中共有的lncRNA有9 260个,陆地棉特有的lncRNA为24 856个,亚洲棉特有的lncRNA为6 076个。为进一步探讨lncRNA在2个棉种中对纤维发育机制的调控异同,对这些共有和特有的lncRNA进行KEGG富集通路分析。发现亚洲棉特有的lncRNA主要参与植物激素信号转导途径、糖类代谢及代谢、苯丙烷生物合成及代谢途径;2个棉种共有的lncRNA主要参与花生四烯酸代谢、植物激素信号转导和内黄酮生物合成途径等;陆地棉共有的lncRNA主要参与肌醇磷酸盐代谢、脂质代谢、氨基酸降解、蛋白质加工和角质、软木质和蜡质生物合成途径等(图9)。
3 讨论
纤维起始和快速伸长阶段是影响棉花纤维产量和质量的关键步骤。因此,了解棉纤维发育过程中的各种分子途径至关重要。在过去的10年里,通过遗传学筛选,挖掘出许多调控棉纤维发育的基因[46]。然而,调控胚珠表皮分化和纤维伸长过程的机制目前还尚不清楚。本研究系统地比较分析了2个短纤维性状差异显著的亚洲棉lncRNA。通过分析胚珠和纤维起始、分化和伸长阶段的转录组数据,鉴定到一批与短纤维发育相关的新lncRNA。发现差异lncRNA的靶基因主要富集在植物激素信号转导和蛋白质加工过程。建立了lncRNA-mRNA调控短纤维起始和形成的网络图,首次发现大量lncRNA在短纤维发育阶段特异性表达。这些数据为进一步研究lncRNA调控棉花纤维发育提供了良好的基础。
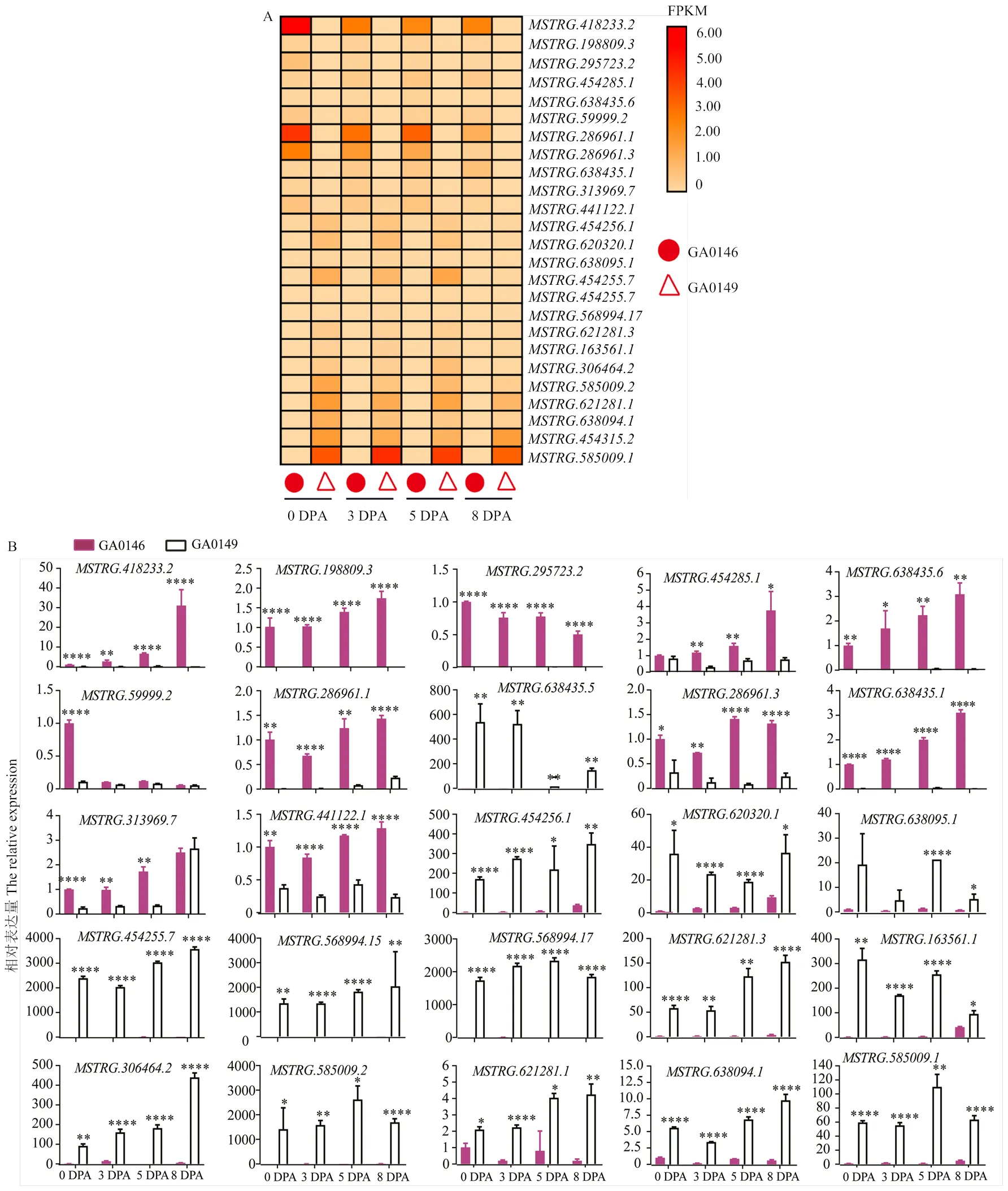
A:热图显示亚洲棉野生型(GA0146)和突变体(GA0149)lncRNA的转录差异;B:25个lncRNA在亚洲棉野生型和短纤维突变体的荧光定量结果,平均值±SD(n=3),*P<0.05,**P<0.01,****P<0.0001
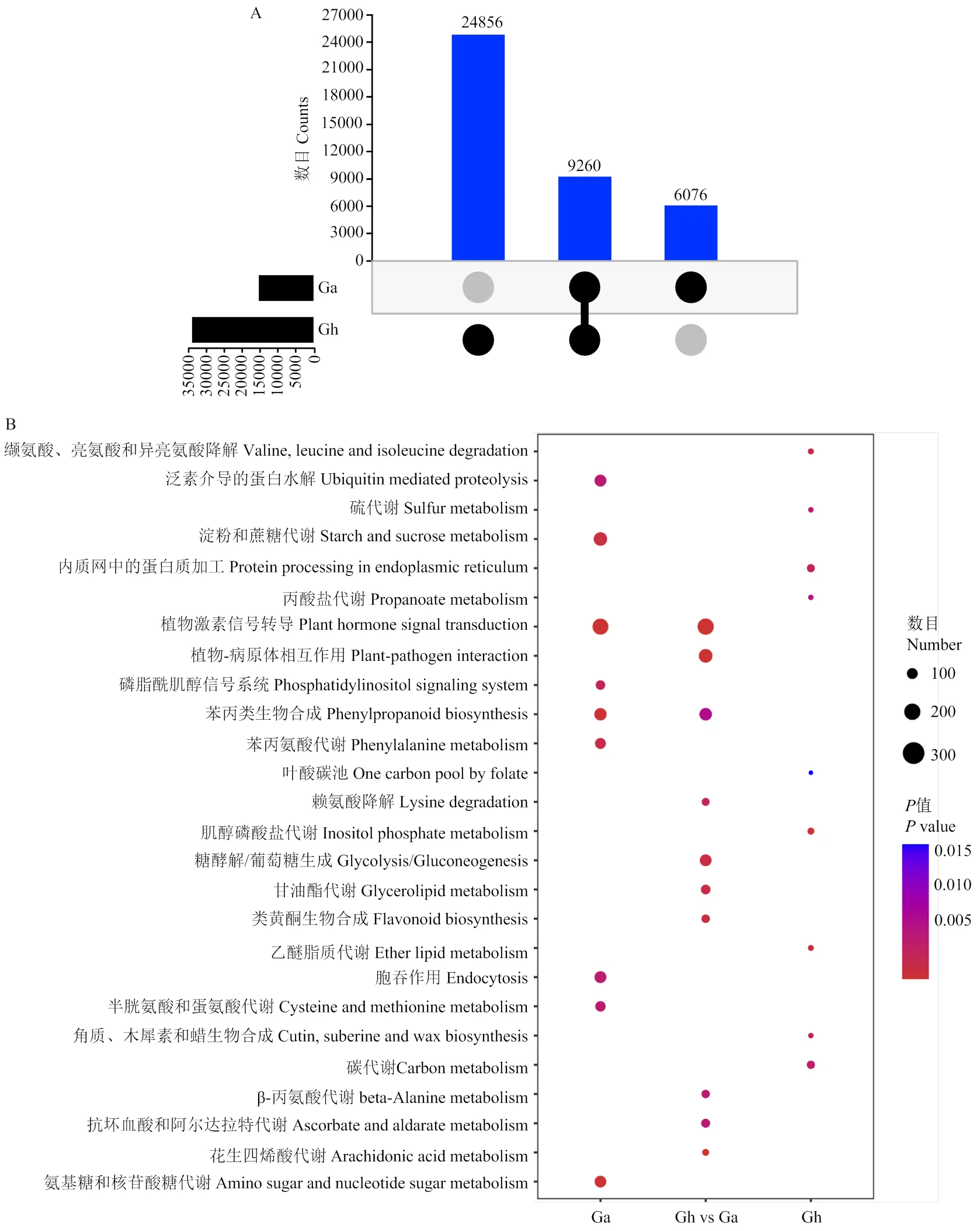
A:lncRNA同源基因在亚洲棉(Ga)和陆地棉(Gh)中的分布;B:lncRNA同源基因在亚洲棉和陆地棉中的富集通路比较
3.1 亚洲棉短纤维发育过程中的lncRNA鉴定
目前,lncRNA已经在许多物种中鉴定出来,在植物生长发育和逆境胁迫过程起重要作用。lncRNA通过调控基因的甲基化水平进而影响水稻的育性[47],参与拟南芥的非生物胁迫和低温春化过程[21],参与玉米的干旱胁迫和与赤霉素相关的植物激素途径[48]。此外lncRNA也参与棉花干旱和盐胁迫等非生物逆境过程以及棉纤维的发育过程[31, 49-50]。上述结果说明,lncRNA是植物中参与转录调控过程的一类重要媒介。另外,有研究报道,在转录水平上,lncRNA通过干扰编码mRNA和非编码RNA的转录、干扰蛋白质结合复合物的形成,通过顺式作用元件调节邻近基因的表达;在转录后水平上lncRNA通过降解mRNA、调控翻译后修饰和可变剪接过程进而影响植物的生长发育。尽管越来越多的报告表明lncRNA在哺乳动物和植物发育过程中发挥作用,但在植物中鉴定的lncRNA尚处于起步阶段,仅有少部分lncRNA被明确鉴定在调节植物发育过程中起重要作用[39]。本研究通过对亚洲棉4个纤维发育时期的RNA-Seq数据进行分析,鉴定出15 339个lncRNA,大部分lncRNA为基因间lncRNA(lincRNAs)。为了研究亚洲棉中lncRNAs的结构特点,对lncRNA和蛋白编码基因(protein coding genes,PCGs)外显子长度、数目、GC含量和表达水平进行分析,结果发现,与编码基因相比,lncRNA含有较低的GC含量、较长的外显子长度和较低的表达水平[35]。本研究序列结构分析发现了同样的结果,lncRNA序列长度短于编码基因的序列长度,lncRNA中的GC含量低于mRNA中的GC含量。lncRNA主要通过顺式和反式作用机制调控相关基因表达,反式作用的lncRNA可以作为染色质的信号,远距离地调节相同染色体域甚至不同染色体中的靶基因表达;顺式作用lncRNA位于编码蛋白的上游和下游,与启动子或共表达基因的顺式核心元件相互作用,从而在转录或转录后水平调节基因表达。本研究根据lncRNA与mRNA的物理位置和表达关系预测了其潜在的顺式和反式靶基因,发现大部分lncRNA对其靶基因的调控方式为顺式调节。Zheng等[35]同样在亚洲棉中发现许多lncRNA以顺式作用方式调控邻近编码蛋白基因的表达。上述结果说明亚洲棉lncRNA具有独特的结构特点。
3.2 差异表达lncRNA在亚洲棉纤维发育过程中的作用
通常认为lncRNA在各种物种中具有高度的组织特异性,lncRNA的这一特性可能意味着它们可以在维持组织特性及组织发育和分化中发挥重要作用。为了进一步了解lncRNA对亚洲棉野生型和短纤维突变体材料的短纤维发育影响,分析了这两个材料4个短纤维发育早期的基因表达谱。结果显示,大部分差异表达的lncRNA表现出组织特异性表达趋势,暗示在纤维发育过程中lncRNA出现了功能分化。研究报道,lncRNA的表达具有时空特异性、高度保守性,在棉花胚珠发育过程中起重要作用[51]。本研究发现、和等仅在野生型的胚珠中特异性表达,在突变体中不表达,说明这些lncRNA对短纤维的起始可能起正调控作用。和在突变体胚珠中的表达水平显著高于其在野生型中的表达水平,说明这两个lncRNA可能抑制亚洲棉短纤维的起始。迄今为止,大多数lncRNA的功能还未完全解析。通过分析lncRNA对编码基因的调控方式,构建lncRNA-mRNA共表达网络,以进一步鉴定lncRNA与mRNA之间的相互调控关系,有助于lncRNA的功能预测[52]。大部分lncRNA与mRNA属于一对多的调控方式,同样的一个编码基因也可以受多个lncRNAs的调控。例如,、和正调控其下游靶基因的表达,进而影响短纤维的发育。上述结果说明lncRNA可能通过抑制或激活其下游靶基因的表达,进而影响亚洲棉短纤维发育过程。但是这些lncRNA的功能研究需要进一步通过转基因和病毒诱导的基因沉默等试验验证。
3.3 参与植物激素信号转导过程的lncRNA调控亚洲棉短纤维的发育
植物激素生长素、乙烯、赤霉素和茉莉酸甲酯在纤维发育过程起重要作用。近年来,许多研究表明,植物主要生长素吲哚-3-乙酸(IAA)通过在纤维细胞中建立浓度梯度来决定棉花纤维的起始[53]。在纤维起始阶段,利用花结合蛋白基因7()启动子驱动IAA生物合成基因在棉花中特异性表达,显著增加了转基因株系胚珠表皮中IAA的积累和起始纤维细胞数量,进而使棉纤维的产量得到了提高[54]。吲哚乙酸诱导蛋白16基因()在野生型的胚珠中表达量非常低,而在棉纤维突变体()开花后的胚珠中表达量迅速升高,说明该基因可能负调控纤维的起始和伸长[55]。本研究通过对差异lncRNA的靶基因进行功能富集发现,大部分靶基因都汇集到植物激素信号转导通路,吲哚乙酸合成酶基因()和生长素响应蛋白基因()在短纤维突变体5 DPA的表达量高于野生型中的表达量,说明这两个基因可能负调控短纤维的起始。茉莉酸甲酯(JA)在纤维发育过程也发挥着一定作用,例如当用JA的抑制剂GhJAZ2在棉花中超表达后,显著抑制了长纤维和短纤维的起始和伸长[56]。向体外离体培养的胚珠中加入2.5 mmol·L-1的JA,能显著抑制纤维的伸长,而当加入0.05 mmol·L-1的JA时,纤维长度显著增加。说明体外离体培养时,高浓度的JA能抑制纤维起始,而适当低浓度的JA能促进纤维伸长。这与本研究结果相符,如参与JA途径的转录因子MYC4()和茉莉酸甲酯合成酶基因JAR1()在突变体5DPA表达量高,8 DPA表达量低。通过对棉花胚珠中ABA含量进行测定发现,长纤维突变体开花当天(0 DPA)胚珠中ABA的含量高于野生型胚珠中的ABA含量,说明ABA可能作为一个负调节因子参与棉纤维的发育[57]。本研究发现ABA信号因子受体ABI2()在突变体5 DPA中的表达量显著高于在野生型中的表达量,这与前人结果相符合。但是上述结果需要进一步的试验证明。
3.4 lncRNA调控亚洲棉和陆地棉纤维机制的异同
异源四倍体栽培种棉陆地棉(,A1A1D1D1)和海岛棉(,A2A2D2D2)是由二倍体A基因组棉(亚洲棉或者草棉)与二倍体D基因组(雷蒙德氏棉)通过天然杂交加倍而形成。后来又经历大约五千年的独立驯化和人工选择,以及环境适应,形成现代棉花栽培品种[58]。二倍体棉种和四倍体棉种的棉纤维性状不相同,亚洲棉纤维粗短且产量低,陆地棉具有纤维品质好、产量高和适应性强等特点,是主要的栽培种。为解析异源四倍体陆地棉和二倍体亚洲棉纤维发育机制的异同。Zhao等[59]从表观遗传学方面对亚洲棉、雷蒙德氏棉、陆地棉及由亚洲棉和雷蒙德氏棉杂交F1的胚珠和叶片中的lncRNA鉴定,发现80% lncRNA在异源四倍体棉花基因组中等位表达,基因组冲击会产生新的非编码转录本。通过对二倍体亚洲棉和四倍体陆地棉lncRNA进行鉴定,显示由二倍体进化到四倍体的过程可能由于基因组重排产生了新的lncRNA[60]。本研究通过对2个棉种lncRNA进行富集通路分析发现,亚洲棉及2个棉种共有的lncRNA主要富集在植物信号转导途径,陆地棉lncRNA主要参与脂肪酸合成代谢途径。说明由于地理位置的不同及基因组重组和自然选择的关系,在多倍化形成过程中lncRNA产生了新的功能分化。
总之,利用生物信息学方法,鉴定出许多与亚洲棉花纤维发育相关的lncRNA,为未来研究lncRNA在棉花纤维中的调控作用和功能分析提供了数据基础。未来的工作将旨在解析lncRNA与棉花纤维发育相关的生物学功能及其提高纤维产量和品质的遗传学机制。
4 结论
从亚洲棉短纤维突变体(GA0149)和其野生型(GA0146)4个短纤维发育早期(0、3、5和8 DPA)的转录组数据中共鉴定出15 329个lncRNA和1 932个具有差异表达的lncRNA。同时,发现lncRNA的GC含量低于mRNA的GC含量、序列长度小于mRNA、表达方式具有组织特异性。单个lncRNA可以同时调控多个靶基因的表达,而单个mRNA也可以受多个lncRNA的调控。差异表达的lncRNA通过调控植物激素信号转导通路基因的表达,影响亚洲棉短纤维的发育。大部分差异表达的lncRNA的靶基因都汇集到植物激素信号转导通路,其中,吲哚乙酸合成酶基因()和生长素响应蛋白基因()可能抑制纤维的起始。
[1] KIM H J, TRIPLETT B A. Cotton fiber growth in planta and in vitro. Models for plant cell elongation and cell wall biogenesis. Plant Physiology, 2001, 127(4): 1361-1366.
[2] STEWART J M. Fiber initiation on the cotton ovule (). American Journal of Botany, 1975, 62(7): 723-730.
[3] WU H T, TIAN Y, WAN Q, FANG L, GUAN X Y, CHEN J D, HU Y, YE W X, ZHANG H, GUO W Z, CHEN X Y, Zhang T Z. Genetics and evolution of MIXTA genes regulating cotton lint fiber development. The New Phytologist, 2018, 217(2): 883-895.
[4] WAN Q, GUAN X Y, YANG N N, WU H T, PAN M Q, LIU B L, FANG L, YANG S P, HU Y, YE W X, ZHANG H, MA P Y, CHEN J D, WANG Q, MEI G F, CAI C P, YANG D L, WANG J W, GUO W Z, ZHANG W H, CHEN X Y, ZHANG T Z. Small interfering RNAs from bidirectional transcripts ofregulate cotton fiber development. The New Phytologist, 2016, 210(4): 1298-1310.
[5] CHEN W, LI Y, ZHU S H, FANG S T, ZHAO L J, GUO Y, WANG J Y, YUAN L, LU Y J, LIU F, YAO J B, ZHANG Y S. A retrotransposon insertion inis likely responsible for the lintless locus liof tetraploid cotton. Frontiers in Plant Science, 2020, 11: 593679.
[6] NAOUMKINA M, THYSSEN G N, FANG D D, LI P, FLORANE C B. Elucidation of sequence polymorphism in fuzzless-seed cotton lines. Molecular Genetics and Genomics, 2021, 296(1): 193-206.
[7] ZHU Q H, STILLER W, MONCUQUET P, GORDON S, YUAN Y M, BARNES S, WILSON I. Genetic mapping and transcriptomic characterization of a new fuzzless-tufted cottonseed mutant. G3 Genes|Genomes|Genetics, 2021, 11(1): jkaa042.
[8] DING M Q, CAO Y F, HE S E, SUN J, DAI H Q, ZHANG H, SUN C D, JIANG Y R, PATERSON A H, RONG J K., a candidate gene for theSMA-4 mutant, promotes trichome and fiber initiation by cellular H2O2and Ca2+signals. Plant Molecular Biology,2020, 103(4/5): 409-423.
[9] LIU X Y, MONCUQUET P, ZHU Q H, STILLER W, ZHANG Z S, WILSON I. Genetic identification and transcriptome analysis of lintless and fuzzless traits inL.. International Journal of Molecular Sciences, 2020, 21(5): 1675-1696.
[10] DU S J, DONG C J, ZHANG B, LAI T F, DU X M, LIU J Y. Comparative proteomic analysis reveals differentially expressed proteins correlated with fuzz fiber initiation in diploid cotton (L.). Journal of Proteomics, 2013, 82: 113-129.
[11] WANG X Y, MIAO Y C, CAI Y F, SUN G F, JIA Y H, SONG S, PAN Z E, ZHANG Y M, WANG L Y, FU G Y, GAO Q, JI G X, WANG P P, CHEN B J, PENG Z, ZHANG X M, WANG X, DING Y, HU D W, GENG X L, WANG L R, PANG B Y, GONG W F, HE S P, DU X M. Large-fragment insertion activates gene() and is associated with the fuzz and trichome reduction in cotton (). Plant Biotechnology Journal, 2021, 19(6): 1110-1124.
[12] FENG X X, CHENG H L, ZUO D Y, ZHANG Y P, WANG Q L, LIU K, ASHRAF J, YANG Q H, LI S M, CHEN X Q, SONG G L. Fine mapping and identification of the fuzzless genein DPL972 (). Theoretical and Applied Genetics, 2019, 132(8): 2169-2179.
[13] FENG X X, LIU S, CHENG H L, ZUO D Y, ZHANG Y P, WANG Q L, LV L M, SONG G L. Weighted gene co-expression network analysis reveals hub genes contributing to fuzz development in. Genes, 2021, 12(5): 753-768.
[14] LIU J, JUNG C, XU J, WANG H, DENG S L, BERNAD L, ARENAS-HUERTERO C, CHUA N H. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in. The Plant Cell, 2012, 24(11): 4333-4345.
[15] GUTTMAN M, RUSSELL P, INGOLIA N T, WEISSMAN J S, LANDER E S. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins.Cell, 2013, 154(1): 240-251.
[16] PONTING C P, OLIVER P L, REIK W. Evolution and functions of long noncoding RNAs. Cell, 2009, 136(4): 629-641.
[17] WANG H L V, CHEKANOVA J A. Long noncoding RNAs in plants. Advances in Experimental Medicine and Biology. Singapore: Springer Singapore, 2017, 1008: 133-154.
[18] SUN X, ZHENG H X, SUI N. Regulation mechanism of long noncoding RNA in plant response to stress. Biochemical and Biophysical Research Communications, 2018, 503(2): 402-407.
[19] ZHANG X P, SHEN J, XU Q J, DONG J, SONG L R, WANG W, SHEN F F. Long noncoding RNA lncRNA354 functions as a competing endogenous RNA of miR160b to regulategenes in response to salt stress in upland cotton. Plant, Cell & Environment, 2021, 44(10): 3302-3321.
[20] ZHANG L, LIU J L, CHENG J R, SUN Q, ZHANG Y, LIU J G, LI H M, ZHANG Z, WANG P, CAI C W, CHU Z Y, ZHANG X, YUAN Y L, SHI Y Z, CAI Y F.andmodulate cell wall defense genes to regulate cotton resistance towilt. Plant Physiology, 2022, 189(1): 264-284.
[21] SUN Q W, CSORBA T, SKOURTI-STATHAKI K, PROUDFOOT N J, DEAN C. R-loop stabilization represses antisense transcription at thelocus. Science, 2013, 340(6132): 619-621.
[22] JABNOUNE M, SECCO D, LECAMPION C, ROBAGLIA C, SHU Q Y, POIRIER Y. A rice-natural antisense RNA acts as a translational enhancer for its cognate mRNA and contributes to phosphate homeostasis and plant fitness. The Plant Cell, 2013, 25(10): 4166-4182.
[23] DING J H, SHEN J Q, MAO H L, XIE W B, LI X H, ZHANG Q F. RNA-directed DNA methylation is involved in regulating photoperiod- sensitive male sterility in rice. Molecular Plant, 2012, 5(6): 1210-1216.
[24] ZHANG Y C, LIAO J Y, LI Z Y, YU Y, ZHANG J P, LI Q F, QU L H, SHU W S, CHEN Y Q. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biology, 2014, 15(12): 512.
[25] ZHANG G Y, CHEN D G, ZHANG T, DUAN A G, ZHANG J G, HE C Y. Transcriptomic and functional analyses unveil the role of long non-coding RNAs in anthocyanin biosynthesis during sea buckthorn fruit ripening. DNA Research, 2018, 25(5): 465-476.
[26] YANG T, MA H Y, ZHANG J, WU T, SONG T T, TIAN J, YAO Y C. Systematic identification of long noncoding RNAs expressed during light-induced anthocyanin accumulation in apple fruit. The Plant Journal, 2019, 100(3): 572-590.
[27] YANG Z E, GE X Y, YANG Z R, QIN W Q, SUN G F, WANG Z, LI Z, LIU J, WU J, WANG Y, LU L L, WANG P, MO H J, ZHANG X Y, LI F G. Extensive intraspecific gene order and gene structural variations in upland cotton cultivars. Nature Communications, 2019, 10(1): 1-13.
[28] DU X M, HUANG G, HE S P, YANG Z E, SUN G F, MA X F, LI N, ZHANG X Y, SUN J L, LIU M, JIA Y H, PAN Z E, GONG W F, LIU Z H, ZHU H Q, MA L, LIU F Y, YANG D G, WANG F, FAN W, GONG Q, PENG Z, WANG L R, WANG X Y, XU S J, SHANG H H, LU C R, ZHENG H K, HUANG S W, LIN T, ZHU Y X, LI F G. Resequencing of 243 diploid cotton accessions based on an updated a genome identifies the genetic basis of key agronomic traits. Nature Genetics, 2018, 50(6): 796-802.
[29] WANG M J, YUAN D J, TU L L, GAO W H, HE Y H, HU H Y, WANG P C, LIU N, LINDSEY K, ZHANG X L. Long noncoding RNAs and their proposed functions in fibre development of cotton (). The New Phytologist, 2015, 207(4): 1181-1197.
[30] 闫飞林. 棉花纤维起始相关长链非编码RNA的功能鉴定[D]. 武汉: 华中农业大学, 2019.
YAN F L. The functional identification of long non-coding RNA related to cotton fiber initial[D]. Wuhan: huazhong agricultural university, 2019. (in Chinese)
[31] ZOU C S, WANG Q L, LU C R, YANG W C, ZHANG Y P, CHENG H L, FENG X X, PROSPER M A, SONG G L. Transcriptome analysis reveals long noncoding RNAs involved in fiber development in cotton (). Science China Life Sciences, 2016, 59(2): 164-171.
[32] HU H Y, WANG M J, DING Y H, ZHU S T, ZHAO G N, TU L L, ZHANG X L. Transcriptomic repertoires depict the initiation of lint and fuzz fibres in cotton (L.). Plant Biotechnology Journal, 2018, 16(5): 1002-1012.
[33] SALIH H, GONG W F, HE S P, XIA W, ODONGO M R, DU X M. Long non-coding RNAs and their potential functions in Ligon- lintless-1 mutant cotton during fiber development. BMC Genomics, 2019, 20(1): 661-678.
[34] ZHANG X P, DONG J, DENG F N, WANG W, CHENG Y Y, SONG L R, HU M J, SHEN J, XU Q J, SHEN F F. The long non-coding RNA lncRNA973 is involved in cotton response to salt stress. BMC Plant Biology, 2019, 19(1): 459-475.
[35] ZHENG X M, CHEN Y J, ZHOU Y F, SHI K K, HU X, LI D Y, YE H Z, ZHOU Y, WANG K. Full-length annotation with multistrategy RNA-seq uncovers transcriptional regulation of lncRNAs in cotton. Plant Physiology, 2021, 185(1): 179-195.
[36] KIM D, PERTEA G, TRAPNELL C, PIMENTEL H, KELLEY R, SALZBERG S L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 2013, 14(4): R36.
[37] ANDERS S, PYL P T, HUBER W. HTSeq–a python framework to work with high-throughput sequencing data. Bioinformatics, 2015, 31(2): 166-169.
[38] TRAPNELL C, ROBERTS A, GOFF L, PERTEA G, KIM D, KELLEY D R, PIMENTEL H, SALZBERG S L, RINN J L, PACHTER L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols, 2012, 7(3): 562-578.
[39] DATTA R, PAUL S. Long non-coding RNAs: Fine-tuning the developmental responses in plants. Journal of Biosciences, 2019, 44(4): 77-88.
[40] SUN L, LUO H T, BU D C, ZHAO G G, YU K T, ZHANG C H, LIU Y N, CHEN R S, ZHAO Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Research, 2013, 41(17): e166.
[41] YOUNG M D, WAKEFIELD M J, SMYTH G K, OSHLACK A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biology, 2010, 11(2): R14.
[42] WU J M, MAO X Z, CAI T, LUO J C, WEI L P. KOBAS server: a web-based platform for automated annotation and pathway identification. Nucleic Acids Research, 2006, 34(suppl_2): W720-W724.
[43] 王骁. 陆地棉纤维品质和产量杂种优势转录本的动态表达与调控模式分析[D]. 北京: 中国农业科学院, 2021.
WANG X. Study on dynamic expression and regulation pattern of transcripts in the heterosis of fiber quality and yield ofL[D]. Beijing:Chinese Academy of Agricultural Sciences, 2021. (in Chinese)
[44] LIVAK K J, SCHMITTGEN T D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T))method. Methods, 2001, 25(4): 402-408.
[45] 王晓阳, 王丽媛, 潘兆娥, 何守朴, 王骁, 龚文芳, 杜雄明. 亚洲棉短绒突变体纤维发育及其差异基因表达分析. 作物学报, 2020, 46(5): 645-660.
WANG X Y, WANG L Y, PAN Z E, HE S P, WANG X, GONG W F, DU X M. Analysis of differentially expressed genes and fiber development infuzzless mutant. Acta Agronomica Sinica, 2020, 46(5): 645-660. (in Chinese)
[46] PEI Y. The homeodomain-containing transcription factor, GhHOX3, is a key regulator of cotton fiber elongation. Science China Life Sciences, 2015, 58(3): 309-310.
[47] DING J H, LU Q, OUYANG Y D, MAO H L, ZHANG P B, YAO J L, XU C G, LI X H, XIAO J H, ZHANG Q F. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proceedings of the National Academy of Sciences of the United States of America, 2012, 109(7): 2654-2659.
[48] ZHANG W, HAN Z X, GUO Q L, LIU Y, ZHENG Y X, WU F L, JIN W B. Identification of maize long non-coding RNAs responsive to drought stress. Plos One, 2014, 9(6): e98958.
[49] DENG F N, ZHANG X P, WANG W, YUAN R, SHEN F F. Identification of Gossypium hirsutum long non-coding RNAs (lncRNAs) under salt stress. BMC Plant Biology, 2018, 18(1): 23-37.
[50] ZHANG X P, DONG J, DENG F N, WANG W, CHENG Y Y, SONG L R, HU M J, SHEN J, XU Q J, SHEN F F. The long non-coding RNA lncRNA973 is involved in cotton response to salt stress. BMC Plant Biology, 2019, 19(1): 459-475.
[51] NECSULEA A, SOUMILLON M, WARNEFORS M, LIECHTI A, DAISH T, ZELLER U, BAKER J C, GRÜTZNER F, KAESSMANN H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature, 2014, 505(7485): 635-640.
[52] CUI J, LUAN Y S, JIANG N, BAO H, MENG J. Comparative transcriptome analysis between resistant and susceptible tomato allows the identification of lncRNA16397 conferring resistance toby co-expressing glutaredoxin. The Plant Journal, 2017, 89(3): 577-589.
[53] ZHANG M, ZENG J Y, LONG H, XIAO Y H, YAN X Y, PEI Y. Auxin regulates cotton fiber initiation via GhPIN-mediated auxin transport. Plant and Cell Physiology, 2017, 58(2): 385-397.
[54] ZHANG M, ZHENG X L, SONG S Q, ZENG Q W, HOU L, LI D M, ZHAO J, WEI Y, LI X B, LUO M, XIAO Y H, LUO X Y, ZHANG J F, XIANG C B, PEI Y. Spatiotemporal manipulation of auxin biosynthesis in cotton ovule epidermal cells enhances fiber yield and quality. Nature Biotechnology, 2011, 29(5): 453-458.
[55] HAN X Y, XU X Y, FANG D D, ZHANG T Z, GUO W Z. Cloning and expression analysis of novelfamily genes in. Gene, 2012, 503(1): 83-91.
[56] HU H Y, HE X, TU L L, ZHU L F, ZHU S T, GE Z H, ZHANG X L. GhJAZ2 negatively regulates cotton fiber initiation by interacting with the R2R3-MYB transcription factor GhMYB25-like. The Plant Journal, 2016, 88(6): 921-935.
[57] GILBERT M K, BLAND J M, SHOCKEY J M, CAO H P, HINCHLIFFE D J, FANG D D, NAOUMKINA M. A transcript profiling approach reveals an abscisic acid-specific glycosyltransferase (UGT73C14) induced in developing fiber ofmutant of cotton (L.). PLoS One, 2013, 8(9): e75268.
[58] FLAGEL L E, WENDEL J F, UDALL J A. Duplicate gene evolution, homoeologous recombination, and transcriptome characterization in allopolyploid cotton. BMC genomics, 2012, 13: 302-315.
[59] ZHAO T, TAO X Y, FENG S L, WANG L Y, HONG H, MA W, SHANG G D, GUO S S, HE Y X, ZHOU B L, GUAN X Y. LncRNAs in polyploid cotton interspecific hybrids are derived from transposon neofunctionalization. Genome Biology, 2018, 19(1): 195-212.
[60] ZOU X, ALI F, JIN S, LI F, WANG Z. RNA-Seq with a novel glabrous-ZM24reveals some key lncRNAs and the associated targets in fiber initiation of cotton. BMC Plant Biology,2022, 22: 61-77.
Identification and Expression Analysis of Fuzz Fiber Development Related Long Noncoding RNAs in
WANG Xiaoyang1, PENG Zhen1,2, XING Aishuang1, ZHAO Yingrui1, MA Xinli1, LIU Fang1,2, DU Xiongming1, HE Shoupu1,2
1Institute of Cotton Research of Chinese Academy of Agricultural Sciences/National Key Laboratory of Cotton Biological Breeding and Comprehensive Utilization, Anyang 455000, Henan;2School of Agricultural Sciences, Zhengzhou University/Zhengzhou Research Base, National Key Laboratory of Cotton Biological Breeding and Comprehensive Utilization, Zhengzhou 450001
【Objective】Long non-coding RNAs(lncRNAs) are a group of RNA molecules longer than 200 bp with no protein coding capacity, which are involved in various biological regulatory processes. In this study, we aim to analyze the RNA-sequencing data of twoisogenic lines, a fuzzless mutant (GA0149) and its wildtype (GA0146), to identify the lncRNA involved in early fuzz fiber development, providing a foundation for investigation the mechanism of fiber development. 【Method】We collected 0 DPA, 3 DPA and 5 DPA ovule and 8 DPA ovule and fiber from thefuzzless mutant GA0149 and its isogenic line GA0146 with normal fuzz and lint fibers, were used for RNA-seq to identify lncRNA and predict their target genes. Differentially expressed mRNA (DE-mRNA) and lncRNA(DE-lncRNAs) between the samples were identified. The KOBAS software was used to predict the KEGG enrichment pathways which DE-lncRNAs targets were involved in. To ensure the quality of high-through sequencing, 25 DE-lncRNAs were selected for RT-qPCR detection. 【Result】We identified 15 339 lncRNA-encoding transcripts that 11 595 lncRNAs were located to intergenic regions, 2 428 lncRNAs were classified as antisense lncRNAs, 350 were categorized as intronic lncRNAs and 966 belonged to sense lncRNAs. Compared to mRNAs, lncRNAs in Asian cotton showed shorter exons and lower GC content. Most of lncRNAs had cis-regulatory effects on their neighboring mRNAs. We identified 1 932 differentially expressed (DE) lncRNAs, with 8 134 predicted DE-lncRNA target genes. Further analysis showed that 788 genes (mRNA) were differentially expressed (DE-genes) during four fiber development stages. KEGG enrichment pathways analysis showed that DE-target-mRNAs were mainly enriched in plant hormone signal transduction and protein processing in endoplasmic reticulum. Co-expression network analysis revealed that lncRNA () and its associated target genes showed identical expression trends during four fuzz fiber development stages, while lncRNAs () and its associated target genes showed contrary expression tendency, exhibiting dramatic higher expression in fuzzless GA0149 compared to wildtype GA0146. The results of RT-qPCR analysis confirmed the authenticity of our RNA-seq data.【Conclusion】A total of 26 specifically expressed lncRNAs were identified which related to cotton fuzz fiber development process. We further confirmed that these lncRNAs affected the fuzz fiber development by regulating the expression of indole-3-acetic acid-amido synthetase () and Auxin-responsive protein () in the plant hormone signal transduction pathway.
; fuzzless mutant; long non-coding RNAs; regulation network; RT-qPCR

10.3864/j.issn.0578-1752.2023.23.002
2022-12-30;
2023-03-16
国家自然科学基金(32201875)
王晓阳,E-mail:wangxiaoyang198806@126.comwangxiaoyang198806@126.com。通信作者何守朴,E-mail:heshoupu@caas.cnheshoupu@caas.cn。通信作者杜雄明,E-mail:dxm630723@163.comdxm630723@163.com。通信作者刘方,E-mail:liufcri@163.com
(责任编辑 李莉)
