液滴式数字PCR与实时荧光定量PCR检测什曼原虫的方法比较
2023-12-25陈维琳
陈维琳,侯 杰,王 念,马 莹
利什曼病是由利什曼原虫引起的病媒传播寄生虫病,通过雌性白蛉传播,被世界卫生组织列为被忽视的热带病之一[1]。人类利什曼病主要有3种临床表现形式:内脏利什曼病、皮肤利什曼病和皮肤黏膜利什曼病,由不同虫种感染所致。其中,内脏利什曼病又被称为黑热病,主要由杜氏利什曼原虫和婴儿利什曼原虫引起,是最严重的利什曼病。据世界卫生组织2016年全球内脏利什曼病的流行情况,我国每年有超过100例新发病例,以感染婴儿利什曼原虫为主[2]。
内脏利什曼病的经典诊断方法是通过在显微镜下观察骨髓或组织标本中的圆形或椭圆形无鞭毛体[1]。此种方法对人体有创,检测灵敏度随取材部位以及不同检测人员的经验而有所差异,检出率不理想。目前,有多种血清学检测方法,包括直接凝集试验(the direct agglutination test,DAT)、免疫层析实验(the immuno-chromatographic test,ICT)、酶联免疫吸附实验、免疫荧光检测和免疫印迹检测等[1],这些方法存在一些局限性,如钩状效应、交叉反应以及治愈后的患者体内依然存在抗体,不宜作为疗效监测的指标。并且,我国临床实验室没有基于以上检测方法的试剂盒可用于内脏利什曼病的诊断。因此,建立一种可用于常规诊断和监测疗效的灵敏的内脏利什曼病的实验室检测方法是很有必要的。
国内外许多研究建立了利什曼原虫的实时荧光定量PCR(real-time quantitative PCR,qPCR)检测方法,具有良好的灵敏度和特异性[3-5]。但以qPCR建立的检测方法若要对样本中的病原体进行定量,需要使用已知浓度的标准品进行标准曲线的绘制,这对于利什曼原虫的检测有相当难度。液滴式数字PCR(droplet digital PCR,ddPCR)可产生线性信号,不需要标准曲线的绘制即可实现单分子绝对定量。数字PCR的高灵敏度使其可用于靶标DNA低载量、多样性时的检测。有研究报道数字PCR在某些寄生虫感染检测方面具有明显的潜在优势,特别是疟疾和血吸虫病[6]。已有的结果表明数字PCR检测方法对低载量DNA具有较高的敏感性和特异性,可以在种的水平上对寄生虫进行区分鉴定[7-11]。而对于内脏利什曼病检测方面的研究少之又少。因此,本研究的目的是以ddPCR为平台,建立一种内脏利什曼病检测方法,将其结果同建立的qPCR检测方法进行灵敏度、精密度比较,分析ddPCR在内脏利什曼病检测中的可行性,以期为临床诊断、疗效监测提供依据。
1 材料与方法
1.1 标准质粒的构建 从美国国家生物技术信息中心(National Center for Biotechnology Information,NCBI)选取数条杜氏利什曼原虫小环动基体DNA序列(GenBank: AF399822.1、EU370883.1、KU201297.1、X84844.1、Y11401.1、Y11402.1、Z35275.1),使用SnapGene(6.0.2)进行序列比对,确定约200 bp保守区域,位于Y11401.1的1~200 bp。选择pUC57载体构建标准质粒(Sangon Biotech,Shanghai,China),并与标准序列比对,保证其准确性。
1.2 引物和探针设计 根据保守序列,使用Primer Premier 5 设计引物,在NCBI Primer-BlAST上对引物进行特异性验证。随后使用Primer Express 3.0.1设计探针。所设计的引物和探针的信息见表1,在参考序列的位置见图1。
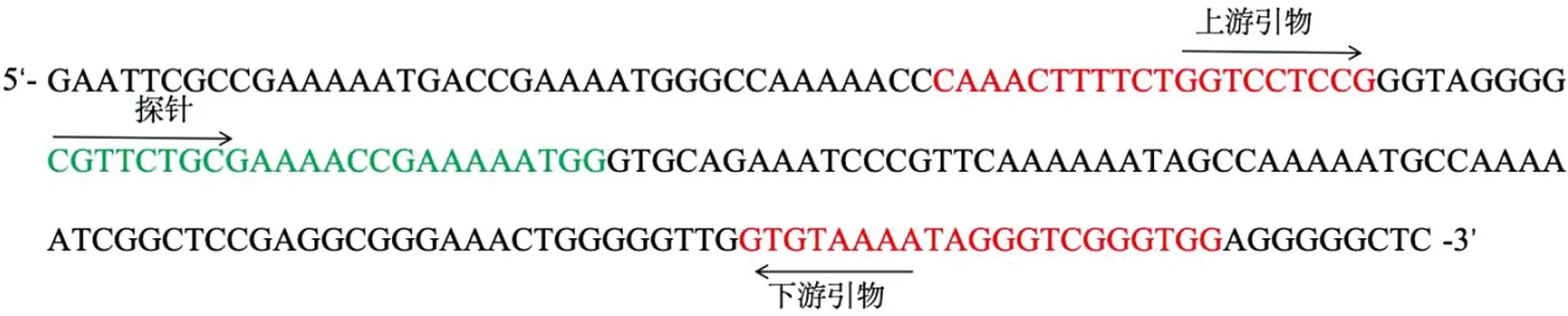
注:红色标记部分为上、下游引物序列,绿色标记部分为探针序列,箭头代表序列方向。图1 引物和探针在参考序列上的位置Fig.1 Positions of the primers and probe on the reference sequence
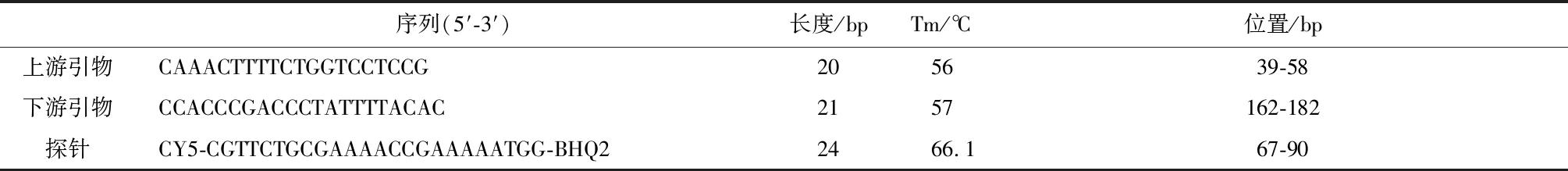
表1 引物和探针Tab.1 Primer and probe information
1.3 实时荧光定量PCR检测方法的建立 以质粒为模板,反应总体系为20 μL:10 μL 2×T5 Fast qPCR Mix(Probe)(Tsingke Biotechnology,Chengdu,China),0.8 μL 10 μmol/L 上游引物,0.8 μL 10 μmol/L下游引物,0.4 μL 10 μmol/L 荧光探针,1 μL模板DNA,7 μL ddH2O。使用Gene9660PCR扩增仪(Bioer Technology, Hangzhou, China)进行扩增。扩增程序为:预变性95 ℃ 5 min后,变性95 ℃ 10 s,退火53 ℃ 20 s,共40个循环,退火时采集荧光。
1.4 液滴式数字PCR检测方法的建立 以质粒为模板,反应总体系为15 μL: 7.5 μL 2×T5 Fast qPCR Mix(Probe)(Tsingke Biotechnology,Chengdu,China),1.35 μL 10 μmol/L 上游引物,1.35 μL 10 μmol/L下游引物,0.375 μL 10 μmol/L 荧光探针,1 μL模板DNA,3.425 μL ddH2O。将总反应体系全部转移至微流控芯片中,随后将芯片转移至DG32 droplet generator(Pilot Gene Technologies, Hangzhou, China),生成液滴,再进行PCR扩增。扩增程序为:预变性95 ℃ 10 min后,变性95 ℃ 30 s,退火53 ℃ 1 min,共40个循环;彻底延伸25 ℃,10 min,反应终止。接下来,将扩增后的芯片加载到iScaner5 Droplet Reader(Pilot Gene Technologies, Hangzhou, China)上,自动测量每个液滴的荧光振幅,使用GenePMS Version 1.0.1(Pilot Gene Technologies, Hangzhou, China)计算靶标DNA的浓度,并以copies/ μL表示。
1.5 qPCR和ddPCR灵敏度、线性及精密度评价 使用5个质粒浓度(分别为1.0×104copies/μL、1.0×103copies/μL、1.0×102copies/μL、1.0×10 copies/μL、1.0 copies/μL)以及阴性对照进行灵敏度测试。使用4个质粒浓度(1.0×104copies/μL、1.0×103copies/μL、1.0×102copies/μL、1.0×10 copies/μL)进行线性和精密度的评价,每个浓度重复10次。
1.6 不同利什曼虫株的检测
1.6.1 DNA提取 将6株液氮冻存的利什曼原虫(表2)解冻,转移至15 mL无菌离心管,加入约5 mL生理盐水,震荡混匀,3 000 r/min,离心10 min,去上清液,再重复此步骤2次。在沉淀中加入蛋白酶K约100 μL,再加入ddH2O补至约400 μL,吹打混匀,放至56 °C温箱孵育过夜。
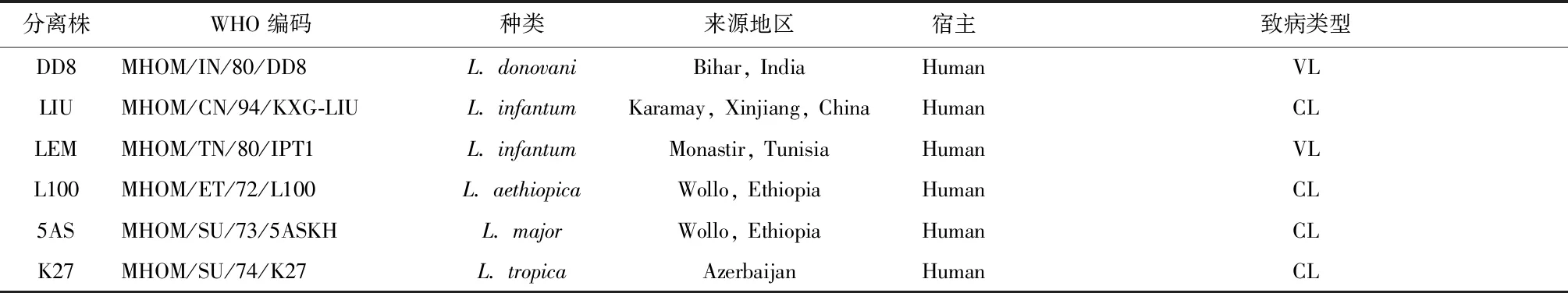
表2 6株利什曼原虫虫株信息Tab.2 Information on six Leishmania isolates
按照动物基因组DNA提取试剂盒(Tsingke Biotechnology,Chengdu,China)操作步骤提取基因组DNA。最终得到50 μL体积的基因组DNA,-20 ℃保存备用。
1.6.2 对已提取的6株利什曼原虫DNA分别进行qPCR和ddPCR检测 以梯度稀释的质粒建立标准曲线,将qPCR检测得到Cq值转换成kDNA拷贝数,用于和ddPCR比较。
2 结 果
2.1 灵敏度和重复性比较 由检测不同梯度浓度的质粒(1.0×104copies/μL、1.0×103copies/μL、1.0×102copies/μL、1.0×10 copies/μL、1.0 copies/μL)所得到的2种方法检测下限结果见图2和图3,qPCR和ddPCR 2种方法最低可检测利什曼原虫质粒拷贝数均为1.0×10 copies/μL。
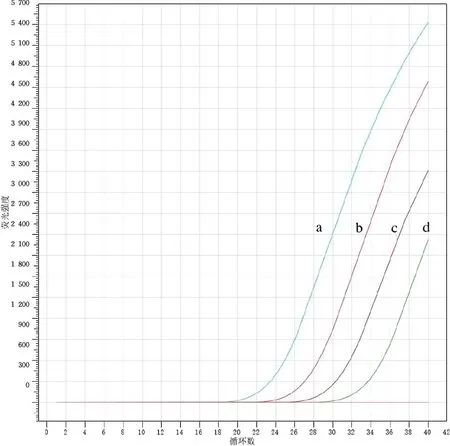
注:质粒浓度a为1.0×104 copies/μL;b为1.0×103 copies/μL;c为1.0×102 copies/μL;d为1.0×10 copies/μL,1.0 copies/μL和阴性对照无扩增。图2 qPCR扩增曲线Fig.2 qPCR amplification curve
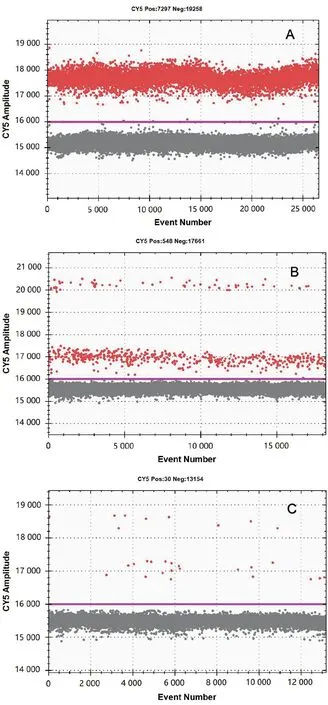
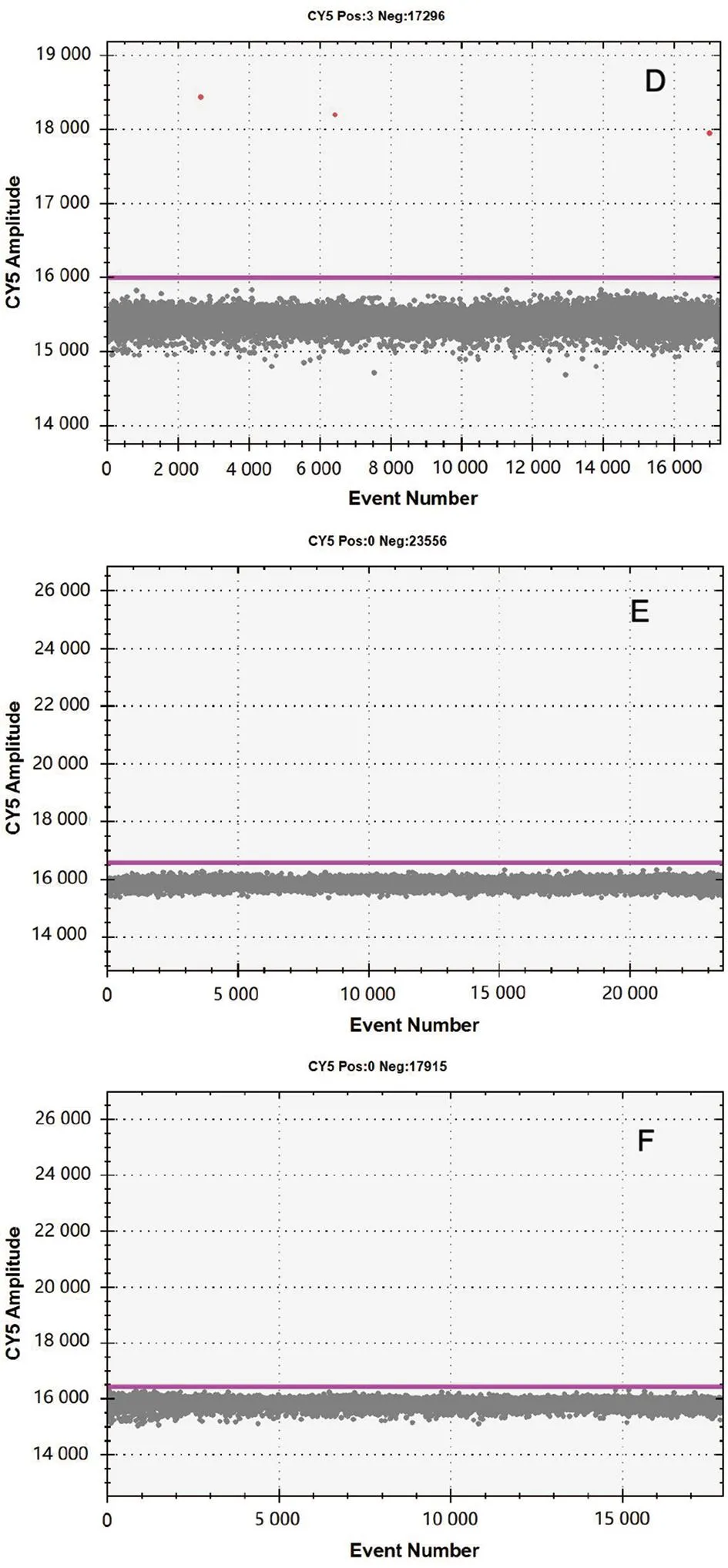
注:质粒浓度A为1.0×104 copies/μL;B为 1.0×103 copies/μL;C为 1.0×102 copies/μL;D为1.0×10 copies/μL;E为1.0 copies/μL;F为阴性对照。
使用不同梯度浓度的质粒(1.0×104copies/μL、1.0×103copies/μL、1.0×102copies/μL、1.0×10 copies/μL)进行线性与精密度评价,每个浓度重复10次。ddPCR的重复检测结果见图4,结果显示ddPCR检测的定量结果与质粒浓度高度相关:R2=0.999 9。从表3可以看出,qPCR的平均Cq值依次为23.63、26.971、30.454、33.761。ddPCR的平均浓度依次是492.458 copies/μL、46.138 copies/μL、3.21 copies/μL、0.25 copies/μL。建立的qPCR检测不同浓度质粒时的变异系数较小(2.96%~4.26%),不同浓度之间的变异系数差别不大,而ddPCR检测的变异系数随着检测样本浓度的降低,变异系数有增大的趋势(12.07%~62.96%)。
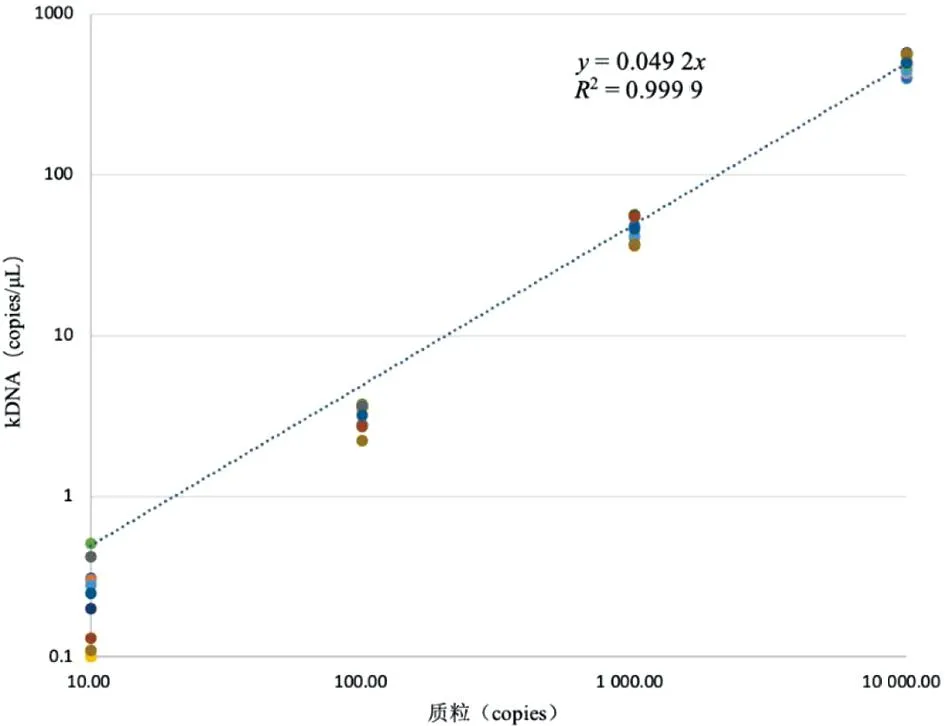
图4 ddPCR平台上10倍稀释质粒与kDNA拷贝数之间的关系Fig.4 Relationship between 10-fold diluted plasmid and kDNA copy number on the ddPCR platform

表3 ddPCR与qPCR重复性比较Tab.3 Comparison of reproducibility between ddPCR and qPCR
2.2 2种方法检测不同利什曼原虫虫株结果比较
对6株利什曼原虫进行qPCR和ddPCR检测。根据不同梯度浓度质粒的拷贝数和Cq值绘制标准曲线(见图5),计算6株利什曼原虫的qPCR定量结果(copies/μL)。2种检测在不同虫株之间的灵敏度具有一定的差异,见表4。qPCR和ddPCR对DD8和L100两株的灵敏度相近[15.81vs17.78 (copies/μL);1.25vs1.49 (copies/μL)];LIU和LEM两株的qPCR检测结果高于ddPCR[99.76vs28.68 (copies/μL);3.97vs0.38 (copies/μL)],qPCR检测方法对其更加灵敏;5AS和K27两株的ddPCR检测结果高于qPCR[18.82vs1.25 (copies/μL);1.61vs0.4 (copies/μL)],ddPCR对其更加灵敏。L100和5AS两株qPCR检测结果接近,但是ddPCR检测结果相差很大。

图5 qPCR标准曲线Fig.5 Standard curve for qPCR
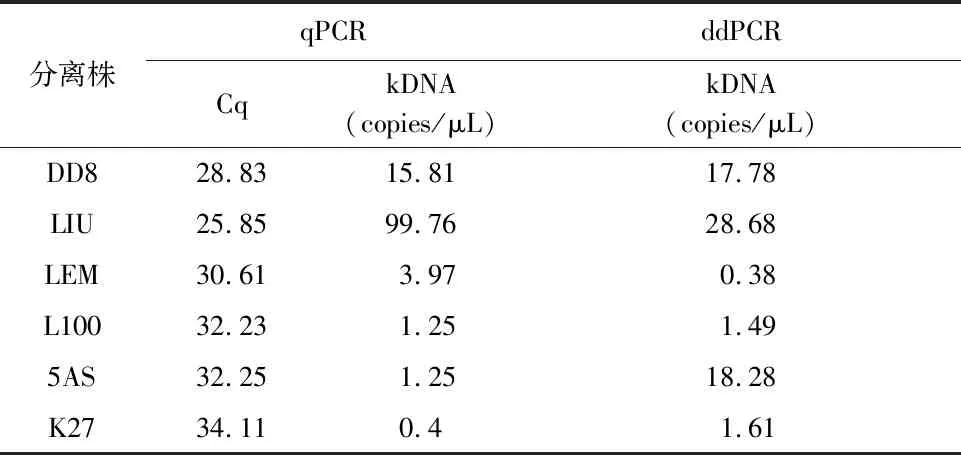
表4 qPCR和ddPCR检测不同利什曼原虫虫株结果对比Tab.4 Comparison of the results of qPCR and ddPCR for detection of Leishmania isolates
3 讨 论
目前临床上内脏利什曼病的诊断方法有显微镜检查、血清学检测、分子生物学技术等。显微镜检查需要观察到无鞭毛体,其在巨噬细胞内繁殖,导致巨噬细胞大量破坏和增生,巨噬细胞增生主要见于脾脏、肝脏、淋巴结和骨髓等器官,检测敏感性和取材部位有关:脾脏90%以上,骨髓50%~80%以上,淋巴结吸出物甚至更低[12]。除了在合并艾滋病毒感染的患者中有较高的寄生虫血症外,血液样本的敏感性很低[13]。国外文献报告有几种血清学检测方法可用,最常用的是基于杜氏利什曼原虫全前鞭毛体的DAT和基于rK39的ICT,但特异性抗体可在患者体内长时间存在,不能区分现症感染和既往感染,即不能作为监测治疗效果的指标[14-15],并且这些产品国内很难获得。分子技术具有较高的灵敏度和特异性,目前国内外已有许多研究建立了qPCR技术检测利什曼原虫。芦晶等[16]将建立的qPCR检测方法与rK39检测方法对比,qPCR方法展示了较好的灵敏度和特异度;另一项研究[17]表明,qPCR检测方法可以作为准确检测内脏利什曼病、黑热病后皮肤利什曼病以及复发内脏利什曼病患者静脉血或皮肤病变组织中利什曼原虫的一种分子工具, 并且能够评估治疗效果。已报道用于利什曼原虫检测的分子靶标有kDNA、18SrDNA、7SLRNA、内部转录间隔区(ITS)、迷你外显子(mini-exon)、HSP70、GP63等[18]。由于kDNA的高拷贝数(约10 000拷贝/寄生虫细胞),其广泛作为利什曼原虫检测的分子标志物[19-22]。
本研究中我们选择了利什曼原虫的小环动基体DNA作为分子靶标,针对其约200 bp的保守片段设计引物和探针,建立了qPCR和ddPCR 2种方法。qPCR定量需要用已知浓度的标准品建立标准曲线,在本研究中,由于利什曼原虫的标准品难获得,故而以质粒构建标准曲线。通过Cq值换算得到的利什曼原虫kDNA拷贝数与实际值之间存在一定的差距。ddPCR的原理是将总PCR反应体系分隔在几万个纳升级的液滴中,形成“油包水“的结构,然后进行PCR反应,含有单分子模板的液滴在扩增后具有荧光信号,当仪器读取每一个微滴荧光信号后,根据荧光信号和泊松分布原理可以计算出样品的浓度,实现单分子绝对定量[23-25]。需要注意的是,本研究中ddPCR实际检测的质粒拷贝数低于预期值。可能是由于预期值是通过已知分子量的DNA换算得来,有一定的误差,并且在质粒稀释过程中存在损耗。
本研究以质粒作为样本,和qPCR检测方法相比,ddPCR检测方法在灵敏度上并没有表现出显著的优势,检测下限均为1.0×10 copies/μL。同样的,从仅有的一篇关于利什曼原虫ddPCR检测方法的研究中发现[26],以L18S为靶标,将稀释后的巴西利什曼原虫作为标准品,确定最低检测限,结果显示ddPCR平台的最低检测限远高于已发表的实时荧光定量PCR平台的检测限(100 vs 1个寄生虫/mL)。在其他寄生虫研究中,ddPCR在低浓度标本的检测中表现出优势,可作为布病的分子流行病学调查的有效手段,以及样本中布鲁氏菌浓度的确定在指导临床用药、监测病情发展和观察疗效中具有重要意义[27]。因此,后续还需要更多的研究探讨如何优化体系,从而提升利什曼原虫ddPCR检测方法的灵敏度。
在本研究中,ddPCR的重复性随着样本浓度的降低,数值波动越大。主要考虑以下几点原因:不同于qPCR,ddPCR得到的结果是每个反应样品浓度(copies/μL),不同样品浓度之间的变化会大于以Cq值呈现出来的变化;在实际操作过程中,液滴式数字PCR转液以及压片不均匀会造成损耗,当样品浓度降低,损耗带来的影响增大。在Ramírez JD等[26]的研究中,在高浓度利什曼原虫样本中,ddPCR的精密度弱于qPCR,随着利什曼原虫浓度的降低,二者的变异系数越接近。
从检测导致内脏利什曼病的虫株结果来看,L.infantum(LIU和LEM)的qPCR检测方法比ddPCR检测方法更加灵敏,而二者对L.donovani(DD8)检测灵敏度差异不大。考虑提取的虫株kDNA与构建质粒的序列之间的相似程度未达到100%,引物、探针和靶标的结合程度会影响扩增效率。和qCPR相比,ddPCR对反应的条件(反应预混液、探针、温度、时间等)要求更高,对ddPCR扩增效率影响更大。通过NCBI Primer-BLAST验证,本方法的引物和探针未见其可扩增其他种类的虫株,从实验结果上看,二者也可检测导致皮肤利什曼病的虫株,其中ddPCR在检测L.major上表现出了较好的灵敏度。对于qPCR检测结果相似的L.aethiopica(L100)和L.major(5AS),ddPCR检测结果却表现出较大的差异。因此,可能需要重新寻找靶标设计引物和探针,探索qPCR和ddPCR在除L.infantum和L.donovani外虫株之间的检测优缺点。
对目标DNA进行检测和绝对定量的ddPCR方法与qPCR方法相比,不需要建立标准曲线,可以更直观地得到待测样品的浓度。除此之外,由于ddPCR将总体系生成上万个油包水的有效的样本分区,避免了非特异性扩增以及最小化或消除液滴间的交叉污染[28]。但是,ddPCR检测方法具有以下局限性,检测通量小;所需的仪器设备以及芯片价格较为昂贵;增加的转液、生成液滴、阅片步骤对操作人员要求较高,导致耗费的时间较长。与qPCR相比,样品浓度过高(超过105copies/μL)可能会影响有效液滴的生成,这样会导致检测结果与实际结果不符,因此对于高浓度样本需要稀释后进行检测。因此,以杜氏利什曼原虫kDNA为靶标,建立的ddPCR检测方法用于临床还需进行验证。
本研究首次以杜氏利什曼原虫小环动基体DNA为分子靶标,根据其保守序列设计引物和探针,尝试建立液滴式数字PCR检测方法,并以质粒为样本从检测限、重复性、成本等方面将其与qPCR进行比较。在灵敏度方面,2种方法的检测下限均为1.0×10 copies/μL;ddPCR的重复性随待测样本浓度降低而越差,而qPCR的重复性更加稳定。因此,和qPCR相比,ddPCR没有表现出特别明显的优势。在对不同利什曼原虫虫株检测中,二者灵敏度具有差异,ddPCR对个别分离株表现出了较高灵敏度。ddPCR所需的检测时间和耗材价格均高于qPCR。数字PCR被认为是寄生虫学中的一种“新”技术,由于高度敏感性和特异性,人们对其临床诊断和筛查的潜力越来越感兴趣,大多数研究主要用来分析检测低寄生虫负荷和区分不同的物种[29]。利什曼原虫ddPCR检测方法的建立,以及在内脏利什曼病的临床诊断方面的应用还需要更多的研究。
利益冲突:无
