Comparison and phylogeny on mitochondrial genome of marine and freshwater taxa of genus Hildenbrandia(Florideophyceae, Rhodophyta)*
2023-12-23FangruNANJuanLIJiaFENGJunpingQiLIUXudongLIUShulianXIE
Fangru NAN, Juan LI, Jia FENG, Junping LÜ, Qi LIU, Xudong LIU, Shulian XIE
School of Life Science, Shanxi University, Taiyuan 030006, China
Abstract Hildenbrandia is an early diverged lineage in Florideophyceae, Rhodophyta.The species diversity of this genus is still unresolved due to the simple morphology and limited molecular information.The mitochondrial genome of freshwater H.jigongshanensis was determined in this study.The freshwater H.jigongshanensis had a larger mitochondrial genome than the marine H.rubra and GC content was higher.Collinear alignment structure was observed between the mitochondrial genomes of H.jigongshanensis and H.rubra, except for one block that was encoded on the complement strand.More introns were found in mitochondrial genome of H.jigongshanensis than in H.rubra, and H.jigongshanensis shares the common feature with Bangiophyceae that two introns were distributed in cox1.Comparison of mitochondrial genome organization suggests that H.jigongshanensis preserves characters that could be hypothetically more similar to the ancestor of Bangiophyceae and Florideophyceae, which differ with previous studies based on chloroplast, and nuclear markers.More mitochondrial genomes and phylogenetic analyses combing nuclear, chloroplast and mitochondrial genomes are needed to clarify this discrepancy.Mitochondrion-based phylogeny in this study resulted in better solution at both the deep and recent derived nodes than single-gene phylogenies.Most protein-coding genes between H.jigongshanensis and H.rubra were identical except atp8, which was present in H.jigongshanensis while absent from H.rubra.This finding follows the trend that high Ka/Ks ratio genes are more frequently lost than low Ka/Ks ratio ones in red algae.
Keyword: red algae; organelle genome; freshwater species; phylogenomics
1 INTRODUCTION
Red algal genusHildenbrandiaNardo belongs to subclass Hildenbrandiophycidae, an early derived lineage in Florideophyceae that inhabits in both marine and freshwater environments (Yang et al.,2016).The morphology ofHildenbrandiais characterized by simple crustose thallus construction with a single basal layer and multiple derived vertical rows of cells.The marine and freshwater species resemble each other in vegetative morphology but differ in mode of reproduction (Sherwood et al.,2002).Freshwater species reproduce by gemmae or fragmentation, whereas marine species reproduce by tetrasporangia or fragmentation.Thallus construction of this genus was simple and the reproductive structures that are often used in red algal classification, differ between marine and freshwater species.Therefore, traditional systematics of this genus was controversial.More molecular and morphological studies on Asian freshwaterHildenbrandiaspecimens are needed to clarify the taxonomy and more studies are needed to clarify existence of already described species based on morphological character only (Vieira et al., 2021a,b).It was reported that freshwaterHildenbrandiaspecies in Europe are monophyletic whereas those in North America are paraphyletic among the marine species (Sherwood and Sheath, 1999, 2000,2003; Sherwood et al., 2002; Vieira et al., 2021a).Consequently, there exists taxonomy in dispute among samples from different continents based on molecular analysis because phylogenies based on few markers are not well resolved.
With the increasing development of nextgeneration sequencing technologies, red algal organelle genome information is more accessible(Yang et al., 2015; Lee et al., 2018; Oliveira et al.,2018; Paiano et al., 2018; Cho et al., 2020).Up to now, 109 mitochondrial genomes were accessible in the NCBI website (https://blast.ncbi.nlm.nih.gov/).The number is still inadequate considering the total number of red algae and there are still some taxonomic groups unrepresented.The mitochondrial genomes proved highly conserved among multicellular red algae of the Florideophyceae (Yang et al., 2015).In contrast, extensive mitochondrial gene rearrangements were reported between Bangiophyceae and Florideophyceae and up to 29 lineage-specific genes were found to be absent in some classes and present in others (Yang et al.,2015).Therefore, more mitochondrial genomes are needed to further evaluate the evolution of mitochondrial genome structure in unexplored groups.
Hildenbrandiophycidae is the earliest diverging subclass in class Florideophyceae.The overall gene synteny is conserved among subclasses in Florideophyceae, except for the Hildenbrandiophycidae,suggesting the distinctiveness of this subclass (Yang et al., 2015).Despite its evolutionary significance,the genome information of this lineage is rare with only two chloroplast genomes and one mitochondrial genome reported (Yang et al., 2015;Lee et al., 2016).The unique mitochondrial genome information was from marineH.rubrawhile the information of freshwater representatives of genusHildenbrandiais absent (Yang et al., 2015).Mitochondrial genome based phylogeny could provide novel insights into the relationships among diverse taxonomic units and was useful for comparisons at species or population in Rhodophyta(Iha et al., 2018).Mitochondrial genome data from major representatives could be a promising tool to further resolve the relationships of the red algae.
The current study presents mitochondrial genome fromHildenbrandiajigongshanensisF.Nan & S.Xie, a freshwater species firstly reported from China and widely distributed in China and Japan (Nan et al., 2017a, 2019; Vieira et al., 2021b).The features of mitochondrial genome ofH.jigongshanensiswere compared with that of marineH.rubraand phylogenetic tree based on mitochondrial genomes was reconstructed.Additionally, the mitochondrial genome sequenced in this study provides more evidences for the red algal phylogenetic research.
2 MATERIAL AND METHOD
2.1 Specimen collection and preparation
Algal specimens ofH.jigongshanensiswere collected from Niangziguan Lake, Shanxi Province,China (37°58′24.6″N, 113°53′27.9″E) on July 7,2016.The specimens were transferred to laboratory as soon as possible and stored at -80 °C after quick freezing using liquid nitrogen.
2.2 Genome sequencing, assembly and annotation
Total DNA was extracted from preserved thalli following the protocol described by Saunders (1993)with modifications following Vis and Sheath (1997).Two paired-end sequencing libraries were constructed using the Illumina Hiseq 2 500 with insertion fragments of 350 bp.After quality control of the Illumina-generated reads, clean data were denovo assembled using SPAdes 3.8.2 (Bankevich et al., 2012).The assembled contigs were blast with other Rhodophyta mitochondrial genomes and the matched contigs were screened out and extended by a baiting and iteration method using the Price software (Ruby et al., 2013).Extended contigs were loaded as reference sequences using Bowtie 2.1.0 and the mapped reads were assembled again using SPAdes 3.8.2 (Langmead and Salzberg, 2012).The final mitochondrial genome was obtained as a complete circular structure.
The putative Open-Reading Frames (ORFs)were predicted by Unipro UGENE v.38, and protein-coding genes were annotated using blastp(Okonechnikov et al., 2012).Potential protein encoding segments were searched in the newly sequenced DNA and predicted proteins were verified using Smart Blast of the ORF finder.The ribosomal RNA genes were identified by comparing the known genomes of related species on the NCBI website (https://last.ncbi.nlm.nih.gov/).Transfer RNAs were identified using the tRNAscan-SE Search Server6 (http://lowelab.ucsc.edu/tRNAscan-SE/).A graphical representation of the annotated genome was drawn with OGDRAW (http://ogdraw.mpimpgolm.mpg.de/cgi-bin/ogdraw.pl).The newly sequenced mitochondrial genome was submitted to GenBank under the accession number OM981155.
2.3 Structure comparison of mitochondrial genomes
The collinear comparison of mitochondrial genomes between marine and freshwaterHildenbrandiawas executed.Additionally, the collinear comparisons between genusHildenbrandiaand florideophycean genusPalmariaand bangiophycean genusPyropiawere executed by Mauve ver.2.3.1 using default settings (Darling et al., 2004).
2.4 Phylogenomic analysis
Red algal mitochondrial genomes of 25 species were downloaded from NCBI according to the blasting results (https://www.ncbi.nlm.nih.gov/genbank/) (Supplementary Table S1).Mitochondrial genome ofH.jigongshanensiswas aligned with other 25 Rhodophyta mitochondrial genomes using Mauve ver.2.3.1 under the progressive mode(Darling et al., 2004).The resulting file was used to extract syntenic alignments by HomBlocks (Bi et al., 2018).The orthologous sequences at the genome-scale generated by HomBlocks were used to construct phylogenetic trees.Modeltest was used to infer the optimal evolutionary models of the dataset (Posada and Buckley, 2004).The Neighborjoining method was performed in the MEGA 5.0 with 1 000 bootstrap repetitions (Tamura et al.,2011).Maximum likelihood tree was constructed using RaxML ver.8 under 1 000 bootstrap replicates(Felsenstein, 1981; Stamatakis, 2014).Bayesian inference was run for 1 100 000 generations with sampling every 100 generations under the temperature of 0.2.After discarding the first 25% of trees as burnin, posterior probabilities were calculated using MrBayes 3.2 (Rannala and Yang, 1996; Ronquist et al., 2012).Mitochondrial genome length, GC content and other features (length of CDS, rRNA, tRNA,intron, non-coding region) of each taxa in the phylogenetic tree were calculated and the features were labeled on the tree using online tools EvolView(http://www.evolgenius.info/evolview.html).
2.5 Functional gene loss and substitution rates calculation
The key functional genes were searched in each taxon of the phylogenetic tree.The non-synonymous and synonymous substitution rates (Ka/Ks) for each gene were calculated using the KaKs_calculator(Wang et al., 2010), and the settings were as follows: genetic code table 4; method of calculation:YN.The Ka/Ks values generated for each gene were displayed into the Box-plot using OriginPro 2016(OriginLab Corporation USA).
3 RESULT
3.1 Feature of mitochondrial genome of Hildenbrandia jigongshanensis
The mitochondrial genome ofH.jigongshanensiswas 42 459 bp in length and GC content was 30.22%.The encoding regions were 26 112 bp in length.The complete mitochondrial genome encoded 52 genes consisting of 38 protein coding genes (including 16 ORF), 12 tRNAs and 2 rRNAs,among which 22 genes were encoded by the heavy strand (H-strand) and 30 genes were encoded by the light strand (L-strand) (Fig.1).Seven introns were found inH.jigongshanensismitochondrial genome.Two introns were found in the genescox1 andnad5 and one intron was found in genesnad2,nad4, andcox2.The tRNAs found inH.jigongshanensisranged from 72 to 83 bp and represented 2.13% of the complete sequence.Out of the 12 tRNA genes,11 tRNA genes were located on the L-strand.Both the large and small subunit ribosomal RNA were located on the H chain, representing 7.17% of the total sequence.The lengths of large and small subunit ribosomal RNAs were 1 719 bp and 1 325 bp respectively and GC contents were 29.61% and 32.30%, respectively.
3.2 Synteny analysis and structure comparison
The mitochondrial genome ofH.jigongshanensis(42 459 bp) was larger thanH.rubra(33 066 bp)with longer genes and non-coding regions (Table 1).Collinear alignment structure was observed between the mitochondrial genomes ofH.jigongshanensisandH.rubra, except for one block was encoded on the complement strand (Fig.2a).InH.jigongshanensis, the rearranged block encoded genes includingtrnM,trnE,trnN,trnU,nad6,rps12, andsecY.When compared with the florideophycean genusPalmaria, 4 blocks (14 genes corresponding tocob,cox3,nad1,nad3,trnL,trnG,sdh2,sdh3,trnF,trnQ,trnS,trnG,trnP, andatp9)were rearranged and when compared to the bangiophycean genusPorphyra, 5 blocks (14 genes corresponding tonad1,nad3,trnL,trnG,sdh2,sdh3,trnF,trnQ,trnS,trnG,trnP,atp9,secY, andcox2) were rearranged inH.jigongshanensis(Fig.2b).Most homologous blocks were rearranged betweenH.jigongshanensisand bangiophycean genusPorphyra, which was similar withH.rubra.There are 17 genes rearranged betweenH.rubraandPorphyraumbilicaliswhereas 14 genes were rearranged betweenH.jigongshanensisandPo.umbilicalis.Seven genes were rearranged betweenH.rubraandPalmariapalmataand 14 genes were rearranged betweenH.jigongshanensisandPa.palmata.Fewer genes were rearranged betweenH.jigongshanesisand Bangiophyceae compared withH.rubra(14 vs.17), whereas more genes were rearranged betweenH.jigongshanesisand other Florideophyceae thanH.rubra(14 vs.7).
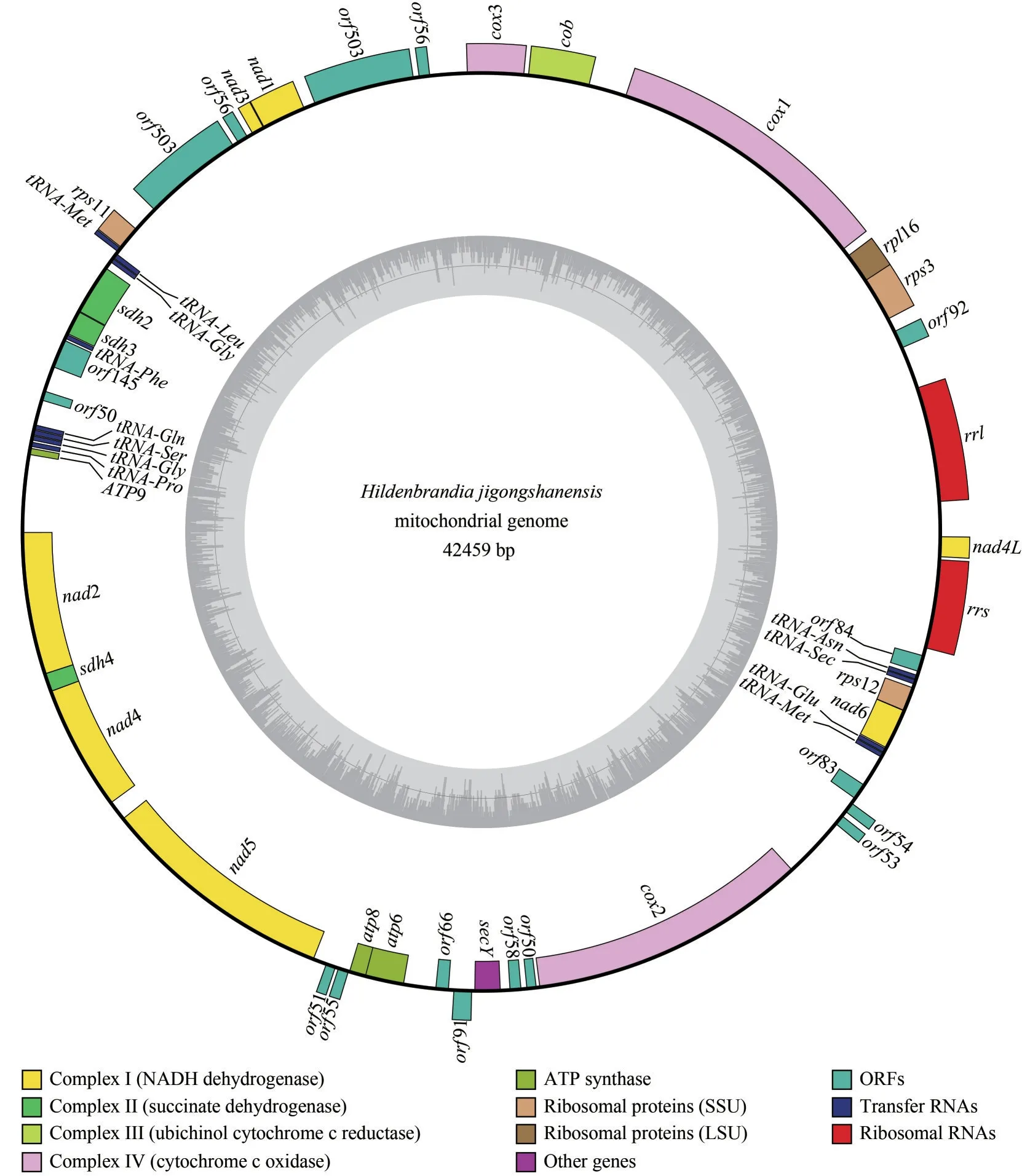
Fig.1 Mitochondrial genome map of Hildenbrandia jigongshanensis
3.3 Phylogenetic analysis
Phylogenetic trees based on orthologous sequences extracted from mitochondrial genomes constructed using the Bayesian Inference, the maximum likelihood, and the neighbor-joining methods are consistent in topological structures.Thus, only the maximum likelihood tree is illustrated with supporting values of ML bootstrap, the Bayesianposterior probabilities and neighbor-joining labeling on the nodes of the tree (Fig.3).With Cyanidiophyceae setting as outgroup, the phylogenetic tree consisted of three main branches including Compsopogonophyceae, Bangiophyceae, and Florideophyceae.With Compsopogonophyceae resolved as the earliest divergent lineage, the classes Bangiophyceae and Florideophyceae were resolved as sister with high support (99.4/1/97).Bangiophyceae and Florideophyceae formed an independent branch supported by full values and 98.1/1/99, respectively.In the Florideophyceae clade, the subclass Hildenbrandiophycidae as the earliest diverged lineage and subclasses including Nemaliophycidae, Rhodymeniophycidae, and Corallinophycidae clustered together (61.2/0.96/99).In Nemaliophycidae subclass, the freshwater order Batrachospermales and Thoreales clustered together with the marine member of Palmariales supported by 99.1/1/93.In Hildenbrandiophycidae subclass,H.jigongshanensisandH.rubrawere resolved in a fully supported clade.

Table 1 Mitochondrial genomic features of each taxon in the phylogenetic tree

Fig.2 Synteny alignment of mitochondrial genomes among different lineages of Rhodophyta
The mitochondrial genomes of Florideophyceae lineage were generally longer than the Bangiophyceae (Fig.3).In Florideophyceae lineage,the freshwater members including generaThorea,Sheathia,Sirodotia, andKumanoain subclass Nemaliophycidae were slightly smaller than the marinePalmariain mitochondrial genome size.The subclass Hildenbrandiophycidae owned the largest mitochondrial genomes among class Florideophyceae(Fig.3).
The percentage of the mitochondrial genome that represents CDSs in Hildenbrandiophycidae(45.6%–52.2%) was lower than that of other subclasses in Florideophycidae (62.8%–73.6%).Similarly, the percentages of rRNA and tRNA in mitochondrial genome of Hildenbrandiophycidae(7.2%–10.1% and 2.1%–4.1% respectively) were lower than those of other subclasses in Florideophycidae (11.8%–16.4% and 4.6%–7.2%respectively).Whereas the percentages of intron sequences in mitochondrial genomes of Hildenbrandiophycidae (7.1%–16.9% and 2.1%–4.1%,respectively) were higher.The introns were absent in most members of other Florideophyceae subclasses except forPalmariapalmata(3.0%)andParalemaneasp.(7.5%).Percentage of noncoding regions in mitochondrial genome of Hildenbrandiophycidae (21.6%–33.1%) was higher than that of other subclasses in Florideophycidae(4.8%–16.7%).In Hildenbrandiophycidae, percentages of CDS and introns in mitochondrial genome ofH.jigongshanensiswere higher than those ofH.rubrawhereas the percentages of tRNA, rRNA and noncoding were lower than those ofH.rubra.
GC contents of the classes Cyanidiophyceae and Compsopogonophyceae exceptCyanidiumchilensewere lower (26.40%–27.38%) than in the class Bangiophyceae.Mitochondrial genomic GC content of Bangiophyceae (30.67%–33.49%) was generally higher than the Florideophyceae (26.94%–32.96%)except forH.rubra(32.20%), genusPalmaria(32.19%–32.96%) andParalemaneasp.(32.42%).In Hildenbrandiophycidae, GC content ofH.jigongshanensiswas lower (30.22%) thanH.rubra(32.20%).
3.4 Encoding genes comparison among closerelated lineages in Rhodophyta

Fig.3 Phylogenetic trees based on mitochondrial genomes, with each genomic features labeled on the tree
Atp4 (synonymous withymf39) was absent in some members of Bangiophyceae.Neitheratp4 norymf39 was found in genusHildenbrandia.Among other subclasses in Florideophyceae, eitheratp4 orymf39 was found.Additionally,atp8 was present in all the members of Bangiophyceae and Florideophyceae exceptH.rubra.Rpl20 was absent from genusHildenbrandiaand most Bangiophyceae members exceptPorphyraumbilicalis(JQ388471),and it was lost in some members of Nemaliophycidae such as genusKumanoaandParalemanea.Additionally,rrn5 was absent from all members of Bangiophyceae and genusHildenbrandia, and it was present in genusKumanoa,Sirodotia,ParalemaneaandSheathiaamong Nemaliophyceae.SecY’(synonymous withtatC) was present in Bangiophyceae and Florideophyceae exceptKumanoa ambigua(MG787095) (Fig.4).
The Ka/Ks calculation results showed that the inatpcomplex, the subunit 4 and 8 were higher in nucleotide substitution rates.The nucleotide substitution rates ofcob,cox, andnadcomplex were rather low.Insdhcomplex, the ratios were high for subunit 3 and 4, but low for subunit 2.The ratios ofrplcomplex were very high especially forrpl20.Inrpscomplex, the ratio of subunit 12 was low but those of subunits 3 and 11 were higher.The substitution ratios ofsecY,ymf39, andtatC were all high and the variance scopes of these three genes were also large.The substitution ratios and variance were the highest inrpl20 and subsequent forsecY (Fig.5).
4 DISCUSSION
The mitochondrial genome ofH.jigongshanensiswas the largest among the florideophycean taxa reported up to now (42.4 kb vs.24.9–33.1 kb), and the GC content was within the range (30.20% vs.23.6%–36.4%) (Yang et al., 2015).Hildenbrandia jigongshanensisinhabited freshwater environments and formed an independent branch with its marine relativeH.rubrabased on multiple analysis (Nan et al., 2017a, b; Vieira et al., 2021b).By comparison,the genome size ofH.jigongshanensiswas larger and GC content was lower thanH.rubra.The size variance was similar than in the chloroplast genome while GC content was opposite (Nan et al., 2017b).The lengths of protein-coding region and intron sequence conspicuously differed inH.jigongshanensisandH.rubra.This result was contrary when compared with the chloroplast genome, which showed no significant difference in protein-coding regions (Nan et al., 2017b).Two introns (distributed inrrlandcox1 respectively)were found in mitochondrial genome of the marineH.rubra(Yang et al., 2015).By comparison, seven introns were found inH.jigongshanensis(no intron inrrl, two introns incox1, and introns in other genes).Compared with Bangiophyceae, a reduced intron incox1 was found inH.rubra, a common feature shared in Florideophyceae.However, there exist two introns incox1 ofH.jigongshanensis,which was similar with Bangiophyceae (Yang et al.,2015).Similar withH.rubra, thetrnI gene was absent in freshwaterH.jigongshanensis, supporting the conclusion thattrnI-intron was lost in genusHildenbrandia(Yang et al., 2015).

Fig.4 Protein-coding gene comparisons among closely related species in Bangiophyceae and Florideophyceae
Two large blocks were rearranged between chloroplast genomes ofH.rubraandH.rivularis(Nan et al., 2017b).By comparison, only one small block was rearranged in the mitochondrial genomes ofH.rubraandH.jigongshanensis, which showed higher conservation of mtDNA gene syntheny and was consistent with previous report (Yang et al.,2015).More similarities in mitochondrial genome organization betweenH.jigongshanensisandPorphyraumbilicalis(Bangiophyceae) were found than those betweenH.rubraand Bangiophyceae taxa.Two large conserved regions, those were,nad2-sdh4-nad4-nad5-atp8-atp6 (ANS segment) andymf39-cox3-cox2-cox1 (CY segment) were found between Bangiophyceae and Florideophyceae in previous report (Yang et al., 2015).InH.rubra,ANS segment was conserved whereas CY segment (cox1-cox2-cox3) was inverted in gene order and lostymf39 (oratp4) compared with the bangiophycean taxa.Similarly, ANS segment was also observed inH.jigongshanensis, whereas genes in CY segment were rearranged (cox1-cox3-cox2).The gene rearrangement may have occurred several times in the evolution of mitochondrial genome.Comparison of mitochondrial genome organization suggests thatH.jigongshanensispreserves characters that could be hypothetically more similar to the ancestor of Bangiophyceae and Florideophyceae.Currently, marineHildenbrandiaspecies is considered primitive than freshwater members based on phylogenetic tree constructed using chloroplastrbcL and nuclear 18S rDNA gene sequences (Sherwood et al., 2002; Vieira et al.,2021b).However, our study of the mitochondrial genome differs from results based on chloroplast and nuclear markers.It may be caused by limited taxon sampling in our study or different evolutionary processes of mitochondrial genome from those of chloroplast and nuclear genomes.Further studies using additional genomes and species are needed to clarify this discrepancy.
Three cyanidiophycean, one compsopogonophycean, seven bangiophycean, and fifteen florideophycean taxa were included in our phylogenetic analysis, thus resulting in a database of 26 mitochondrial genomes from red algae for the phylogenetic and comparative analyses.The result was consistent with multiple-gene based and chloroplast genome based phylogenies (Yoon et al.,2006; Le Gall and Saunders, 2007; Nan et al.,2017b).Mitochondrion-based phylogeny in this study resulted in better solution at both the deep and recent derived nodes by providing high supports at many nodes than phylogenies based on few markers (Nan et al., 2017a, 2019).H.jigongshanensisgrouped withH.rubraand diverged earliest in the Florideophyceae clade,which supports the ancient evolutionary origins ofHildenbrandiain this class (Yang et al., 2016).
Except for the outgroup Cyanidiophyceae, the mitochondrial genome size increased first and then decreased from earlier (Compsogonophyceae) to more recent diverged lineages (Florideophyceae) in red algae, which is consistent with previous reports of both chloroplast and mitochondrial genomes(Nan et al., 2017b).The evolution patterns of mitochondrial genome size diverged among eukaryotic lineages.The evolutionary trend was toward a further compaction of the mitochondrial genome in animals, whereas the mitochondrion genomes tend to increase in size by acquisition of large amount of non-coding DNA in plants (Gray et al., 1998).In the majority of protist, mtDNAs are compact with few or no large non-coding regions(<10% of the mtDNA) (Gray et al., 1998).Surprisingly, non-coding regions accounted for 21.6%–33.1% of the total mtDNA in genusHildenbrandia, which is conspicuously higher than other red algae in our phylogenetic tree(4.7%–16.7%).It has been proved protist mtDNAs have evolved in the direction of higher A+T content generally (Gray et al., 1998).Contrary to the general trend, the GC content of genusHildenbrandia(30.2%–32.2%) was lower than Nemaliophycidae genusPalmaria(32.1%–32.9%).More genomes ofHildenbrandialineage are needed to confirm the observed patterns.
Most protein-coding genes were identical inH.jigongshanensisandH.rubraexceptatp8 that was present inH.jigongshanensiswhile absent fromH.rubra.Theatp8 gene was found in all the other Bangiophyceae and Florideophyceae members, thus suggesting that it was present in the ancestor of genusHildenbrandiaand lost inH.rubra.Pseudogenization or outright loss ofatp8 was observed in the coralline red algaNeogoniolithon spectabileand mitochondrial genomes of parasite species (Hancock et al., 2010; Lee et al., 2018).Theatp8 gene encoded the ATP8 subunit of the F1F0-ATP synthase complex and was reported to have been transferred to the nuclear genome in many lineages including apicomplexans, dinoflagellates,and green algae (Denovan-Wright et al., 1998;Slamovits et al., 2007).Whether theatp8 gene was transferred to the nuclear genome or simply lost inH.rubraremains unclear and further studies are needed.Theatp4 andrpl20 genes were absent in mitochondrial genomes of bothH.jigongshanensisandH.rubra, asymf39 was synonymous withatp4(Burger et al., 2003).The loss ofatp4 was only found in genusHildenbrandiaand occurred only once.The generpl20 was only present in some mitochondrial genomes, and it has been lost inHildenbrandia,Porphyrapurpurea,Bangiafuscopurpurea, the ancestor of sixPyropiaspecies, andWildemania schizophylla(Yang et al., 2015).The mitochondrial genomes of moreHildenbrandiaspecies are needed to elucidate whether this gene might have been lost in the common ancestor of the lineage.
Higher Ka/Ks ratios of theatpandsdhin Rhodophyta mitochondrial genomes have been reported in a previous study (Nan et al., 2017b).Similarly, high Ka/Ks ratios ofatp4,ymf39(synonymous withatp4),atp8,sdh3, andsdh4 were observed in our study.Along with these genes, therplandrpshad high Ka/Ks ratios,especially for therpl20.As aforementioned, loss ofrpl20 was the most common in Bangiophyceae and Florideophyceae taxa to date.Additionally,the absence of the ribosomal and succinate dehydrogenease genes (rpl,rps, andsdh) has been found in land plants (Adams et al., 2002; Adams and Palmer, 2003), and genes includingrpl,rps,sdh,secY, andatp4 have been lost in red algaGaldieria sulphuraria(Cho et al., 2020).Thus, we infer there was a trend that high Ka/Ks ratio genes were more frequently lost than low Ka/Ks ratio ones in red algae.
5 CONCLUSION
The mitochondrial genome of freshwaterH.jigongshanensiswas sequenced and characterized in our study.The size was the largest among the mitochondrial genomes of florideophycean taxa reported up to now, with 42 459 bp in length and GC content of 30.22%.Genome size ofH.jigongshanensiswas larger and GC content was lower than marineH.rubra.The presence of introns was more common inH.jigongshanensisthan inH.rubra.Rearrangement of one small block was observed in the mitochondrial genomes betweenH.rubraandH.jigongshanensis.Compared withH.rubra, fewer genes were rearranged betweenH.jigongshanesisand Bangiophyceae whereas more genes were rearranged betweenH.jigongshanesisand Florideophyceae.Most protein-coding genes betweenH.jigongshanensisandH.rubrawere identical exceptatp8 was present inH.jigongshanensiswhile absent fromH.rubra.Mitochondrion-based phylogeny in this study resulted in better solution at both the deep and recent derived nodes than single-gene based phylogenies.Higher Ka/Ks ratios genes consisting ofatp4,atp8,sdh3,sdh4,rpl, andrpsare more frequently lost than low Ka/Ks ratio ones in red algae.
6 DATA AVAILABILITY STATEMENT
The assembled datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
杂志排行

Journal of Oceanology and Limnology的其它文章
- Characteristics of dissolved sugars in the Southern Yap Trench from sea surface to hadal zone*
- Characteristics of vertical distributions of methane and dimethylsulphoniopropionate in the southern Yap Trench*
- The profile of sound speed and dissolved oxygen in the polymetallic nodules depositional area in the Western Pacific*
- Spatial differentiation and dynamic mechanism of microgeomorphology based on acoustic spectrum data of the Huanghe (Yellow) River Delta*
- Early life history traits of chub mackerel Scomber japonicus in the Oyashio water revealed by otolith microstructure*
- Morphological comparison and gonadotropins cell localization of mature female turbot and mouse pituitary*