结构关系挖掘及其在生物信息中的应用
2023-12-20陈章昭陈未如高胜召
陈章昭,陈未如,张 雪,高胜召,韩 静
(1.沈阳化工大学 计算机科学与技术学院,辽宁 沈阳 110142;2.辽宁省化工过程工业智能化技术重点实验室,辽宁 沈阳 110142)
0 引 言
结构关系挖掘是数据挖掘领域一个全新的分支,是基于序列模式挖掘提出的一种寻找序列模式之间内在结构关系的挖掘方法。该方法将序列模式之间的关系进一步细分,整合成一种由并发、互斥、重复及串行关系组成的复合关系[1-3]。
随着人类基因组计划的启动和高通量测序技术的快速发展,生物医学数据呈现指数增长趋势,面对海量的生物数据,生物信息学的重要性日益凸显[4]。生物信息学的研究内容主要包括发展新的数理信息技术以用于开发生物数据的算法和运用数据挖掘等计算机技术以用于分析解释生物基因信息。如今,应用和开发数据挖掘技术来探索生物系统规律是生物信息学领域最受关注的方向之一,其中包括基因序列分析、蛋白质功能预测、生物进化分析、表达图谱分析等[5-6]。在生物信息领域,结构关系挖掘方法也有重要应用。王翠青等人提出使用支持向量作为提取蛋白质序列中新模式的算法ConSP[7]并进行了并发挖掘。Jing Lu 等人使用真实蛋白质数据集的实验突显了ConSP 方法在蛋白质这种数据中的适用性[8]。现有的结构关系挖掘方法关注的是序列模式间的结构关系,忽略了那些并不是很频繁但却可能有意义的序列,而且在挖掘过程中,主要分析的是序列模式之间而非序列内部的关系,这在面向生物信息挖掘时可能会导致分析得到的结果过于冗余,实际意义不大。因此,本文对现有的结构关系挖掘知识体系做了进一步改进,在原有结构关系挖掘的基础上,改进了并发度、互斥度以及并发关系和互斥关系的概念,基于此提出了面向生物基因信息的结构关系挖掘算法框架。改进后的结构关系挖掘方法将序列之间的结构关系进一步细化到项集之间,并且关注了那些并不频繁但可能存在意义的序列。这样的改动使得在面向生物基因信息挖掘时能够得到更加科学客观的结果,从而确保在研究生物结构、分析生物进化变异等问题时能够快速准确地挖掘到有效的知识。
1 相关问题描述
1.1 有关序列模式和结构关系模式的知识
I={i1,i2, ...,im}是项目的集合,项集是I的非空子集,记为(x1,x2, ...,xk),其中xj∈I,(1 ≤j≤k≤m)。序列S是项集的有序集合,记为{s1,s2, ...,sn},其中每个元素si是一个项集。在事务数据库中,包含S的序列数与事务数据库中的序列总数之比称为序列S的支持度,记为sup(S)。用户指定的最小支持度记为minsup。当序列S的支持度大于等于用户指定的最小支持度,即sup(S)≥minsup 时,则称序列S为频繁序列或序列模式[9]。事务数据库中所有的序列模式构成该数据库的序列模式集,记为SPDB。对于序列S={I1I2...In}和序列S'={I'1I'2...I'n},m<n,如果存在m个正整数1 ≤j1≤j2≤...≤jm≤n,使得Ij1⊆I'j1,Ij2⊆I'j2, ...,Ijm⊆I'jm,则称序列S包含于序列S',记为S⊆S',也称S为S'的子序列或S'为S的超序列[10]。
结构关系模式挖掘是一种基于序列模式挖掘所提出的挖掘任务,旨在寻找隐藏在序列模式间的结构关系,如并发关系模式、互斥关系模式以及重复关系模式等[1-3]。现有结构关系模式挖掘的研究给出了并发度、互斥度、并发序列模式以及互斥序列模式等定义,并在此基础上提出了几种结构关系模式挖掘算法。
1.2 结构关系的相关概念
基因项:基因项ij=<loc, base>由两部分组成, 其中base为生物碱基{A, G, C, T}或蛋白质{G, A, V, L, I, F, W, Y, D, N,E, K, Q, M, S, T, C, P, H, R},loc 为该碱基或蛋白质在所对应序列的绝对位置。
基因序列:由若干个基因项构成的集合称为基因序列,记为{i1,i2, ...,in},其中每个元素ij为一个基因项。
如序列S:{<1, A>, <2, T>, <3, T>, ..., <210, T>, <211, T>,<212, G>, ..., <29 561, T>, <29 562, G>}是由29 562 个基因项构成的新冠病毒基因序列。
并发度:对于序列A={α1,α2, ...,αn},序列数据库SDB中包含A的序列个数与包含A中任意项集的序列个数之比,称为序列A的并发度,记作con(α1,α2, ...,αn),或con(A)。
并发关系:对于序列A={α1,α2, ...,αn},给定客户指定的最小并发度mincon,当con(A)≥mincon 时,称A存在并发关系,表示为[A]=[α1+α2+...+αn]。α1,α2, ...,αn构成一组并发集。特别的,若A为基因序列且并发集中包含n个基因项,则称该并发集为n-基因并发集。
表1 为包含了4 条新冠序列的基因序列数据库GSDB。

表1 基因序列数据库GSDB
若给定最小并发度mincon = 70%, 根据并发度的定义可以得出序列S={<1, A>, <210, T>, <211, T>}的并发度con(<1,A><210, T>, <211, T>)=3/4 ≥mincon, 则称序列S存在并发关系。表示为[S]=[<1, A>+<210, T>+<211, T>]。<1, A>, <210,T>, <211, T>构成一组3-基因并发集。
并发关系具有反单调性:对于给定的序列数据库GSDB,如果序列A={α1,α2, ...,αn}存在并发关系[α1+α2+...+αn],则A的任意一个子序列也存在并发关系。
证明:假设序列A={α1,α2, ...,αn} 且存在并发关系[α1+α2+...+αn],即con(α1,α2, ...,αn)≥mincon,A'为序列A的一个n-1 子序列。在序列库SDB 中,包含A的序列肯定也包含A',即con(A')的分子要大于等于con(A)的分子;由于序列A'较A相比少了一个元素,因此,con(A')的分母要小于等于con(A)的分母。综上可得,con(A')≥con(A)≥mincon,即序列A的任意一个n-1 序列也存在并发关系。以此类推,序列A的任意一个子序列都存在并发关系。
完全并发集:对于并发关系[C1]=[α1+α2+...+αm]和[C2]=[β1+β2+...+βn],m<n。若对∀i(1 ≤I≤m)都存在αi⊆βj(1 ≤j≤n),则称并发关系[C2]包含并发关系[C1]。若基因序列S存在并发关系且不被任意一个并发关系所包含,则称并发关系[S]为完全并发关系,该并发关系的所有基因项构成一组完全并发集。
互斥度:对于序列A={α1,α2, ...,αn},序列数据库SDB中包含且仅包含A中一个项集的序列个数与包含A中任意项集的序列个数之比称为序列A的互斥度,记作xcl(α1,α2, ...,αn)或xcl(A)。
互斥关系:对于序列A={α1,α2, ...,αn},给定客户指定的最小并发度minxcl,当xcl(A)≥minxcl 时,称A存在互斥关系,表示为[A]=[α1⊕α2⊕...⊕αn]。α1,α2, ...,αn构成一组互斥集。特别的,若A为基因序列且互斥集中包含n个基因项,则称该互斥集为n-基因互斥集。
对于给出的GSDB,若给定最小互斥度minxcl =60%,根据互斥度的定义可以得出基因序列S={<29 655,C>, <29 656, A>} 的互斥度为:xcl(<29 655, C>, <29 656,A>)=2/3 ≥minxcl,称序列S存在互斥关系,表示为[S]=[<29 655, C>⊕<29 656, A>]。
根据互斥度与并发度关系,以及并发关系的反单调性质可知,任何一个存在互斥关系的序列A的超序列(包含该A的序列)很容易满足互斥关系,满足这一条件的互斥关系称为平凡互斥关系,这样的互斥关系不是我们关心的,只有那些任意子序列间都存在互斥关系的序列才有意义。
非凡互斥关系:序列A={α1,α2, ...,αn}存在非凡互斥关系,当且仅当A及其所有子序列都满足互斥关系。显然,非凡互斥关系满足反单调性。
关联度:同时包含序列A和B的序列占包含序列A的序列的比例,称为序列A关联B的关联度,记作association(A,B)。
关联关系:对于序列A与B,当A在某一序列中出现时B也有很大概率出现,即A与B的关联度ass(A,B)≥minass(minass 为客户指定的最小关联度),则称序列A与B存在关联关系,表示为[A→B]。
对于给出的GSDB,若给定最小关联度minass =90%,根据关联度的定义可以得出基因序列A={<1, A>, <2, T>, <2,C>}与B={<210, T>, <212, G>}的关联度为:association(A,B)=1.0 ≥minass, 称存在关联关系[{<1, A>, <2, T>, <2,C>}→{<210, T>, <212, G>}]。
2 结构关系挖掘具体过程描述
获取数据集:数据集一般是通过访问资料库、网页抓取和问卷调查手动收集等方式获得。特别的,对于生物基因数据而言,可以从生物基因数据库下载进行研究,如NCBI(https://www.ncbi.nlm.nih.gov/)为美国国家生物技术信息中心,该数据库包含人类基因组、病毒、微生物和新冠病毒等生物基因信息;GISAID(https://db.cngb.org/gisaid/)是全球最大的流感及新型冠状病毒数据平台,该数据库不仅具有最完整的新冠病毒基因组序列数据以及相关临床和流行病学数据,更汇聚了全球诸多科研团队对COVID-19 的研究成果。
预处理:数据预处理是为了提高数据的质量,保证数据的准确性、完整性和一致性。对于生物基因序列而言,从基因数据库下载的基因序列可能存在基因缺失或未知碱基数过多的情况,所以需要将下载的序列进行预处理,去除重复和低质量的序列。其次,虽然同类生物基因序列相似度很高,但序列长度会存在略微偏差,在预处理阶段还需要进行序列对齐操作。
获取变异基因序列组:生物进化的实质是遗传物质的变异。在面向生物信息的研究过程中,由于生物序列和事务序列的特征存在很大差异,如生物序列是由有限个体(碱基或蛋白质)组成的超长序列,且同类生物基因序列的相似性很高[11],因此可以对基因序列的变异点进行针对性研究,这不仅可以很大程度提高挖掘效率,更使得分析生物变异进化过程更加科学、客观。已有研究表明,病毒基因组间的共突变是研究病毒进化的重要标志。例如,Deng Lizong 等人利用氨基酸序列的共突变网络来预测埃博拉病毒的致命性[12]。Olabode E. Omotoso 等人分析发现,新冠病毒序列S蛋白D614G 与其他复发性蛋白共突变对病毒ACE-2 宿主进入产生了影响[13]。Qin Luyao 等人根据SARS-CoV-2 基因组发现了一些共突变模块来推测病毒的进化传播过程[14]。经过预处理后的基因序列长度一致,我们首先选取一条序列作为参考序列,然后将基因组中的序列和选取的参考序列进行序列比对,去除序列中具有一致核苷酸的保守位点,剩余部分则构成了变异基因序列。所有变异基因序列构成变异序列组vGSDB。
挖掘序列间的结构关系:通过结构关系挖掘算法挖掘序列间的结构关系。本文给出了面向生物基因信息的结构关系挖掘算法。
结果可视化表达:由挖掘序列间的结构关系步骤可以得到SDB 结构关系,根据这些结构关系通过相关可视化方法可进一步分析序列库中的信息。如本文根据挖掘得到的新冠序列结构关系生成了GSDB 系统发育框架,从而更好捕获生物基因的进化变异情况。
3 结构关系挖掘算法
由于实验对象选取的是新冠病毒Sars-Cov-2 序列,因此本文在Apriori、Prefixspan 等序列模式挖掘算法的基础上,结合结构关系定义给出了适用于生物信息领域的挖掘算法。
3.1 基于Apriori 的并发关系挖掘算法conApriori
输入:基因序列数据库GSDB,最小并发度mincon
输出:所有的并发集allConcurrentItemSets
算法:
(1)获取GSDB 中所有的基因项T,生成初始候选并发集Lk = T⋈T(k 为2);
(2)令Ck = null; allConcurrentItemSets = null;
(3)do
for each s of Lk
if(con(s)≥mincon)
将s 存入k-并发集Ck 中;
将Ck 存入allConcurrentItemSets;
Lk+1=Ck⋈Ck;
for each c of Lk+1
if(c 存在k-子序列不被Ck 所包含)
将c 从Lk+1 中删除;
while(Lk+1 is not null) ;
3.2 基于PrefixSpan 的并发关系挖掘算法conPrefix
输入:基因序列数据库GSDB,最小并发度mincon
输出:所有的并发集allConcurrentItemSets
算法:
(1)获取GSDB 中所有的基因项T;
(2)令pre = null,preDB = null,prefDBItem = null;
(3)for each s of T
(4)pre = s;
(5)获取前缀pre 对应的投影数据库preDB;
if(preDB is not null)
获取preDB 中的所有基因项preDBItem;
for each c of preDBItem
pre = pre + c;
if (con(pre)≥mincon)
将pre 存入allConcurrentItemSets;
执行步骤(5);
3.3 基于Apriori 的互斥关系挖掘算法excApriori
输入:基因序列数据库GSDB,最小互斥度minxcl
输出:所有非凡互斥集allExclusionaryItemSets
算法:
(1)获取GSDB 中所有的基因项T,生成初始候选互斥集Lk = T⋈T(k 为2);
(2)令Ek = null; allExclusionaryItemSets= null;
(3)do
for each s of Lk
if(xcl(s)≥minxcl)
将s 存入k-非凡互斥集Ek 中;
将Ek 存入allExclusionaryItemSets;
Lk+1=Ek⋈Ek;
for each c of Lk+1
if(c 存在k-子序列不被Ek 所包含)
将c 从Lk+1 中删除;
4 实验
4.1 数据准备和处理
GISAID 数据库按照Pangolin 分类方法将SARS-CoV-2基因组序列划分成了若干个分支。本实验首先将GenBank 中的SARS-CoV-2 参考基因组作为参考序列,再从B 分支,B.1分支以及B.1.126 分支分别选取15 条SARS-CoV-2 基因组序列作为数据集。数据集经数据清洗和序列对齐等预处理操作生成新的基因序列集GSDB。将基因序列集GSDB 中的每条序列和选取的参考序列进行序列比对,去除序列中具有一致核苷酸的保守位点,剩余部分组成变异基因序列。处理得到45 条变异基因序列,构成变异基因序列集vGSDB。
4.2 实验过程及分析
通过conApriori,conPrefix 以及excApriori 算法对变异基因序列集vGSDB 进行结构关系挖掘。
表2 列举了变异基因序列集vGSDB 在最小并发度mincon=0.9 下挖掘得到的部分并发集。由表2 可知,多处核苷酸或氨基酸位点存在高并发变异的情况,如ORF1ab 蛋白的4517H 和S 蛋白的614G 之间满足高并发关系。已有研究表明,SARS-CoV-2 序列产生并发突变可能会对病毒的传播和感染能力产生一定影响,如S 蛋白的L452R 和T478K 并发突变会使刺突蛋白以更高的亲和力附着在ACE2 受体上,从而影响病毒的传 播感染性[15]。

表2 vGSDB 在mincon=0.9 下挖掘得到的部分并发集
图1 表示数据集GSDB 的系统发育情况。在mincon=0.6时,vGSDB 通过算法挖掘得到了15 个完全并发集。构建一个n×m的矩阵,其中n代表选取的45 条样本基因序列,m代表挖掘得到的15 个完全并发集。若基因序列中存在某完全并发集,则将该完全并发集对应位置的值记为1,否则记为0。然后,先将具有相同完全并发集的基因序列放在同一组,再根据组中包含完全并发集的个数将基因序列分配到不同水平。若一个组包含i个完全并发集,则将该组分配到第i级。同时,如果第i级中的完全并发集都被第i+1 级的完全并发集包含,则将第i级的组认定为第i+1 级组的父级。根据该划分规则,样本基因序列集最终被划分为15 个分支。现有一些对SARS-CoV-2 进行分类的方法,如Nextstrain 和Pangoli[16]分别根据分离株和突变的数量以及时空分布的变化来构建系统发育树,从而实现对SARS-CoV-2 的划分。本文提出的根据完全并发集的划分方法可以看作是一种更加精细的划分规则,属于同一分支下的序列亲缘性较高,该方法能够准确有效地识别SARSCoV-2 群体之间的层次关系,这为病毒追根溯源提供了相关依据。

图1 数据集GSDB 的系统发育情况
图2 表示conPrefix 和conApriori 两种挖掘算法在不同相关度下的运行时间效率情况。由图2 可知,随着最小并发度的不断增大,两种挖掘算法所消耗的时间也不断减少。此外,在同一最小并发度下,conPrefix 算法消耗的时间相对较短,效率优于conApriori 算法。
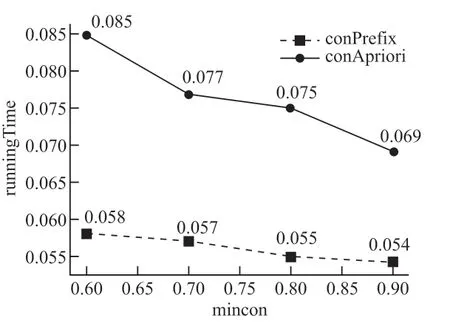
图2 conPrefix 和conApriori 算法在不同并发度下的运行时间变化曲线
图3 表示变异基因序列集vGSDB 通过算法在不同minxcl 下挖掘得到的非凡互斥集个数变化情况。从图3 可得知,在最小互斥度为0.9 时,依然存在着大量的非凡互斥集,如{<1822, I>, <5951, Y>, <69, C>}、{<2611, I>, <5951, Y>,<69, C>, <167, F>}均满足非凡互斥关系,这表明病毒发生了进一步突变,部分序列出现了较新突变位点。

图3 vGSDB 在不同最小互斥度下的非凡互斥集变化曲线
图4 表示变异基因序列集vGSDB 通过算法在不同minass 下挖掘得到的关联关系变化情况。从图4 可得知,在不同的最小关联度下,挖掘得到的关联关系个数相差不大,说明变异序列间的关联性很强,如关联关系{<5951, Y> <73,F>} →{<4715, H> <614, G>},当ORF1ab 蛋白的5951 号位氨基酸突变为Y,E 蛋白的73 号位氨基酸突变为F 时,ORF1ab 蛋白的4715 号位氨基酸突变为H,S 蛋白的614 号位氨基酸突变为G 的可能性很大。
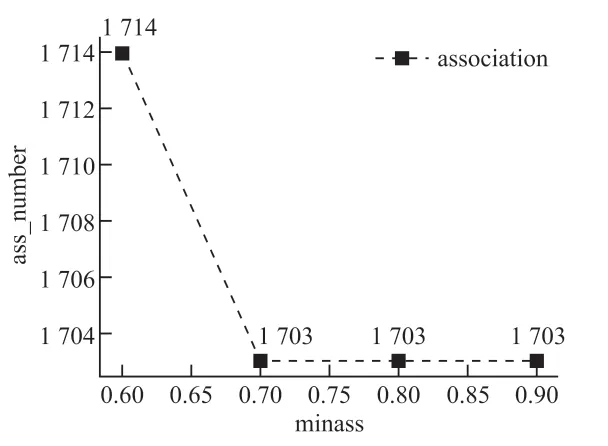
图4 vGSDB 在不同最小关联度下的关联关系变化曲线
5 结 语
在生物信息领域,结构关系是一种普遍存在的关系,如新冠病毒序列S 蛋白D614G 与其他复发性蛋白并发变异会对病毒ACE-2 宿主进入产生影响;癌症患者的基因中,具有互斥关系的基因集合在患者群体中会表现出有且只有一个基因变异的现象;妊娠乳腺癌患者的基因存在关联关系,检测到其中一种修饰,则其它三种也极有可能存在。本文在现有结构关系挖掘方法的基础上做了进一步改进,给出了面向生物信息的结构关系挖掘算法,并将其应用到SARS-CoV-2研究中,通过实验挖掘得到了隐藏在序列集中的并发、互斥和关联关系,由并发关系生成的系统进化树可用于新冠病毒序列间的进化传播关系研究,也进一步验证了挖掘方法的正确有效性。此外,结构关系挖掘还可以应用于诸多场景,合理的应用结构关系挖掘方法对生物基因信息研究具有一定意义。
注:本文通讯作者为陈章昭。
