氯化胆碱-乙二醇深共熔体系电沉积Ni-Mo 合金镀层动力学及其电催化析氢性能
2023-12-18盛施展李文畅王慧华
盛施展,李 林,李文畅,王慧华
(苏州大学 沙钢钢铁学院,江苏 苏州 215021)
氢能源因热值高、零污染等优点而成为重要的清洁能源[1-2],其中,电解水制氢因原料来源广泛、过程清洁环保而备受制氢工业青睐[3-5]。然而,电解水制氢因为电极界面电阻大、能耗高、产氢效率低等缺点,严重制约了其快速发展。贵金属Pt 及其合金因为特殊的能带结构可加快电解水反应速率,提高产氢效率,但因为储量有限及价格昂贵,无法大规模应用,因此,开发低成本、高效的析氢催化材料刻不容缓。根据Engel-Brewer 价键理论,Ni-Mo 合金对于析氢反应具有良好的电催化协同作用[6-7],成为众多研究者关注的一个热点。为进一步提高Ni-Mo 合金电催化析氢性能,研究人员尝试通过调控Ni-Mo 合金成分或优化微观形貌提高其本征活性或表观活性。Zhang 等[8]研究发现,花状微纳米结构的Ni-Mo 合金催化剂具有优异的电催化析氢性能。Zhang 等[9]研究结果表明,Ni-Mo固溶体同样具有良好的析氢催化活性,10 mA·cm-2的析氢过电位仅为60 mV。深共熔溶剂具有特殊的离子结构,利用其作为溶剂电沉积制备Ni-Mo 合金镀层有望对合金形貌及成分进行综合调控,从而实现合金催化性能的大幅提升。Gao 等[10]从氯化胆碱-尿素深共熔体系中电沉积Ni-Mo/Cu 催化材料,其在HER 中的Tafel 斜率为49 mV·dec-1,20 mA·cm-2的过电位仅为63 mV,明显优于水溶液获取的Ni-Mo 合金催化性能[8-9]。此外,Florea 等[11]发现,深共熔体系中电沉积Ni-Mo 合金镀层不仅具有良好的电催化性能,而且与基底具有较好的界面结合力,可以显著提高合金催化稳定性。目前,有关深共熔体系电沉积Ni-Mo 合金镀层的报道较少,其沉积动力学也缺乏系统的研究。众所周知,深共熔溶剂无水或少水,其内部离子结构类型和离子迁移速率与传统水溶液明显不同,铁族金属去极化或原子氢吸附还原等熟知的经典理论已不再适用于深共熔体系[12-14],因此探讨Ni-Mo 合金在深共熔体系中的沉积动力学,尤其合金成分及形貌的演变规律对于制备高性能Ni-Mo 双金属合金催化剂具有重要意义。
基于此,本工作以Cu 片为基体,在氯化胆碱-乙二醇(ChCl-EG)深共熔体系中电沉积制备Ni-Mo 合金镀层,通过交流阻抗谱(EIS)探究Ni-Mo 合金镀层沉积动力学,借助XRD,EDS 以及SEM 分析沉积产物成分及其形貌演变规律,推测电位驱动下的Ni-Mo 合金反应路径,并对不同反应路径产物进行析氢性能表征,旨在理解氯化胆碱-乙二醇深共熔体系中电沉积参数对Ni-Mo 合金镀层电催化析氢性能的影响规律。
1 实验材料与方法
1.1 Ni-Mo 合金镀液配制
实验原料为氯化胆碱(HOC2H4N(CH3)3Cl,ChCl),乙二醇(C2H6O2,EG),六水氯化镍(NiCl2·6H2O),一水柠檬酸(C6H8O7·H2O)和四水钼酸铵(NH4)6Mo7O24·4H2O,所有原料均为分析纯,购于阿拉丁试剂有限公司。将氯化胆碱和乙二醇按照摩尔比1∶2,70 ℃均匀混合后形成无色透明的溶剂(30 mL),然后加入1.209 g 的C6H8O7·H2O,充分搅拌至溶液澄清时加入1.112 g (NH4)6Mo7O24·4H2O,继续搅拌至淡黄色透明溶液,最后加入4.279 g NiCl2·6H2O,搅拌至均匀透明的绿色溶液(Ni-Mo 合金镀液),待用。
1.2 Ni-Mo 合金镀层阻抗测试
将除油、酸洗后的洁净黄铜片作为工作电极,Pt片为对电极,Ag 丝为参比电极,将三根电极垂直置于70 ℃的Ni-Mo 合金镀液中静置15 min,期间不断向电解槽中通入Ar 气,以减少镀液中溶解O2对实验结果的影响,然后进行交流阻抗测试。交流阻抗采用普林斯顿电化学工作站(P3000A),测试频率范围为0.001~10000 Hz,测试电位分别为-0.6,-0.8,-1.0,-1.2,-1.4,-1.5 VvsAg。
1.3 Ni-Mo 合金镀层制备及性能表征
1.3.1 Ni-Mo 合金镀层制备
采用标准三电极体系电沉积制备Ni-Mo合金镀层,其中以处理后的黄铜片(1 cm×1 cm,剩余部分用AB 胶密封)为工作电极,Pt片为对电极,Ag丝为参比电极,沉积温度为70 ℃,沉积时间为5 min,沉积电位参考阻抗测试电位,即-0.6,-0.8,-1.0,-1.2,-1.4,-1.5 V,对应的合金镀层分别命名为Ni-Mo-0.6,Ni-Mo-0.8,Ni-Mo-1.0,Ni-Mo-1.2,Ni-Mo-1.4 和Ni-Mo-1.5。
1.3.2 Ni-Mo 合金镀层性能表征
采用SU5000 型场发射电子显微镜对镀层进行微观形貌分析;借助EDS 能谱仪对镀层成分进行半定量分析;利用X 射线衍射仪(Ultima Ⅳ)对镀层表面进行物相分析;采用UV7504 型紫外-可见分光光度计表征合金镀液的离子类型;利用X 射线光电子能谱(XPS)分析合金镀层表面元素的化学态,靶材为Mg 靶,发射频率为1283.3 eV;采用Fischer-MPO 膜厚仪测量镀层厚度,并计算相应的沉积速率;使用标准三电极体系测试合金镀层电催化析氢性能,其中Hg/HgO/OH-为参比电极,石墨棒为对电极,Ni-Mo 合金镀层为工作电极,电解液为1 mol/L KOH,为方便对比,Ni-Mo 合金电催化析氢测试过程中的电位均换算成可逆氢电极,即PRHE=PHg/HgO/OH-+0.098+0.0591pH。
2 结果与讨论
2.1 Ni-Mo 合金镀层的沉积动力学
图1 为Ni-Mo 合金镀液的紫外-可见吸收光谱和CV 扫描曲线。由图1(a)的UV-Vis 谱图可以看出,Ni-Mo 合金镀液中有四个特征吸收峰,分别位于λ0=218 nm,λ1=396 nm,λ2=656 nm 和λ3=734 nm 处,对比单一(NH4)6Mo7O24和NiCl2溶液的特征吸收峰位置可知,Ni-Mo 合金镀液的特征吸收峰仅为(NH4)6Mo7O24和NiCl2溶液吸收峰的叠加,说明合金镀液中络合离子类型是由镍络合离子和钼络合离子机械混合而成。参考文献[15]可知,200~300 nm 为[Mo7O24(C6H7O7)]5-络合离子特征吸收峰,350~450 nm 为[Ni(EG)3]2+络合离子的特征吸收峰,600~700 nm 和700~800 nm 皆为[NiCl4]2-络合离子的特征吸收峰,进一步说明Ni-Mo 合金镀液中同时存在[Ni(EG)3]2+,[NiCl4]2-和[Mo7O24(C6H7O7)]5-三种络合离子。图1(b)为Ni-Mo 合金镀液在实验电位区间内的CV 曲线,可以看出,Ni-Mo 合金镀液仅表现出一个较宽的还原峰,其起始电位在-0.6 V 处。对比ChCl-EG 溶剂和NiCl2镀液CV 扫描曲线可知,ChCl-EG 溶剂在-0.2~-1.4 VvsAg 没有观察到明显的还原峰,提高极化电位时,仅表现出欧姆降,说明在实验电位范围内ChCl-EG 溶剂是稳定的,而NiCl2镀液在-0.9~-1.05 V 和-1.05~-1.12 V 处分别出现B1和B2两个还原峰。结合图1(a)可知,NiCl2镀液中仅含有[Ni(EG)3]2+和[NiCl4]2-两种络合离子,因此图中B1和B2分别对应于[Ni(EG)3]2+和[NiCl4]2-的还原[16]。很明显,Ni-Mo 合金镀液的还原峰起始电位相比于B1正向移动,说明镀液中的钼酸根离子可以促进镍络合离子的还原。另外,Ni-Mo 合金镀液在较宽的电位区间并未观察到其他明显的还原峰,说明合金中Mo 与Ni可能发生共沉积行为。
图2 为Ni-Mo 合金镀液不同极化电位下的交流阻抗谱图。从图2(a),(b)可以看出,在较低的极化电位(-0.6 V 和-0.8 V)时,整个阻抗谱仅表现出被压扁的容抗弧和典型的扩散弧,并且其变形程度随极化电位增加越发明显,说明在此电位区间镀层呈现多孔态,而且镀层的沉积速率受镀液中离子迁移速度控制。-1.0 V 和-1.2 V 极化电位时(图2(c),(d)),阻抗谱在较低的频率开始出现感抗弧,如频率<0.14 Hz(-1.0 V)或频率<82 mHz(-1.2 V)时均出现明显的感抗弧,且其特征频率随极化电位增加而增大(10 mHz 增大至26 mHz),说明电极表面开始有中间吸附体存在,且中间吸附体数量随极化电位增加逐渐增多。进一步提高极化电位(-1.4 V 和-1.5 V)(图2(e),(f)),可观察到感抗弧减小甚至消失,说明电极表面中间吸附体数量减少或完全被消耗。通常,中间吸附体反应速率受离子扩散速度以及电荷转移速度共同控制,极化电位低,离子扩散速度大于电荷转移速度,中间吸附体可在电极表面积累,因此表现出明显的感抗弧;极化电位高,电荷转移速度大于离子扩散速度,中间吸附体反应消耗快,电极表面积累量减少,感抗弧减小或消失。此外,在高极化电位时较低频率段重新出现新的容抗弧,这可归结为合金镀液中游离出来的H+被还原产生氢气的过程[13]。
图3 为Ni-Mo 合金镀层在不同极化电位下(-0.8,-1.0,-1.2,-1.4 V 和-1.5 V)的SEM 图和EDS分析。可以看出,Ni-Mo 合金镀层的微观形貌受极化电位影响较大,极化电位为-0.8 V 时(图3(a-1),(a-2)),镀层表面较为平整,伴有针刺边缘的粒状颗粒呈现松散堆积,该电位下的合金镀层主要由金属Ni 组成(图3(a-3)),少量的O 可归因于镀层表面吸附氧或金属Ni 氧化所致。当极化电位增加到-1.0 V 时(图3(b-1),(b-2)),镀层表面依旧平整,但颗粒呈现明显的针状结构,说明增加极化电位有利于晶体择优生长,此时合金仍然以金属Ni 为主(图3(b-3))。当极化电位进一步增加至-1.2,-1.4 V 和-1.5 V 时(图3(c-1),(c-2),(d-1),(d-2),(e-1),(e-2)),Ni-Mo 合金镀层表面形貌发生显著变化,呈现出由多个细小晶粒聚集而成的胞状晶,且极化电位越大,胞状晶的晶粒尺寸越大,镀层表面的裂纹越发明显,合金中Mo 含量越高(图3(c-3),(d-3),(e-3)),说明Ni-Mo 合金中的Mo 含量是引起表面形貌改变和镀层内应力提升的重要原因。此外,合金中的O 含量随着极化电位增加先上升后下降,在-1.2 V 时达到最大,结合图2(d)可知,在-1.2 V 时电极表面有大量中间吸附体富集,推断在该电位下[Mo7O24(C6H7O7)]5-开始被还原,生成MoOx等中间吸附体,但随着极化电位增加,中间吸附体还原速度提高,合金中的O 含量又呈现下降趋势(图3(e-3))。

图3 Ni-Mo 合金镀层SEM 照片和EDS 分析(a)Ni-Mo-0.8;(b)Ni-Mo-1.0;(c)Ni-Mo-1.2;(d)Ni-Mo-1.4;(e)Ni-Mo-1.5;(1)低倍;(2)高倍;(3)EDS 分析Fig.3 SEM images and EDS analysis of Ni-Mo alloy coatings(a)Ni-Mo-0.8;(b)Ni-Mo-1.0;(c)Ni-Mo-1.2;(d)Ni-Mo-1.4;(e)Ni-Mo-1.5;(1)low magnification;(2)high magnification;(3)EDS analysis
图4 为不同极化电位下Ni-Mo 合金镀层的XRD 分析。可以看出,在低极化电位时(-0.8 V和-1.0 V),合金镀层表面呈现(111)Ni和(200)Ni衍射峰,且极化电位越大,Ni 衍射峰强度越高,表明结晶形态越好;随着极化电位的增加(-1.2 V),(111)Ni峰形明显宽化,且向低角度微弱偏移,说明有少量的Mo 固溶到Ni 结构中引起晶格膨胀(Mo≈18.6%,质量分数,下同);随着极化电位进一步增加(-1.4 V 和-1.5 V),(111)Ni衍射峰消失,左侧附近出现较小的馒头峰,说明形成具有非晶形态的Ni-Mo 合金。结合图3(e-3)对应的EDS能谱分析结果,可以估算出合金中Ni/Mo 化学计量比约为4,因此Ni-Mo 合金镀层在较高极化电位下可形成Ni4Mo 非晶合金。
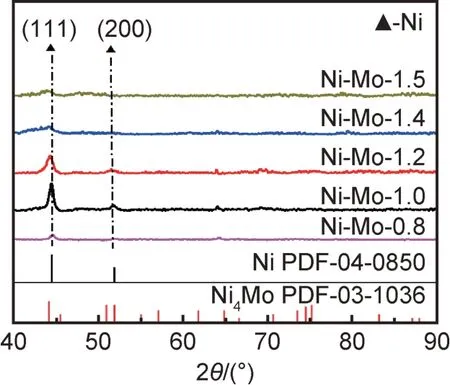
图4 不同极化电位下Ni-Mo 合金镀层的XRD 分析Fig.4 XRD analysis of Ni-Mo alloy coatings fabricated at different polarization potentials
图5 进一步给出Ni-Mo-1.4 合金镀层表面元素化学态分析。从图5(a)可以看出,Ni2p 高分辨图谱可以拟合成五个峰,其中851.98 eV 对应金属态Ni0的特征峰,而位于855.94 eV 和873.51 eV 的两个拟合峰分别对应于Ni—O 的2p3/2和2p1/2自旋轨道特征峰,并伴有典型的卫星峰。Mo3d 图谱中有三个拟合峰(图5(b)),其中结合能为227.7 eV 对应于金属态Mo0特征峰,232.34 eV 和235.49 eV 分别对应于不同价态的Mo—O 键,该结论进一步说明镀层除了Ni-Mo 合金外,还夹杂有NiO 和MoOx,这些氧化物可能归因于活泼金属的表面氧化,该分析结果与EDS 分析结果基本保持一致。
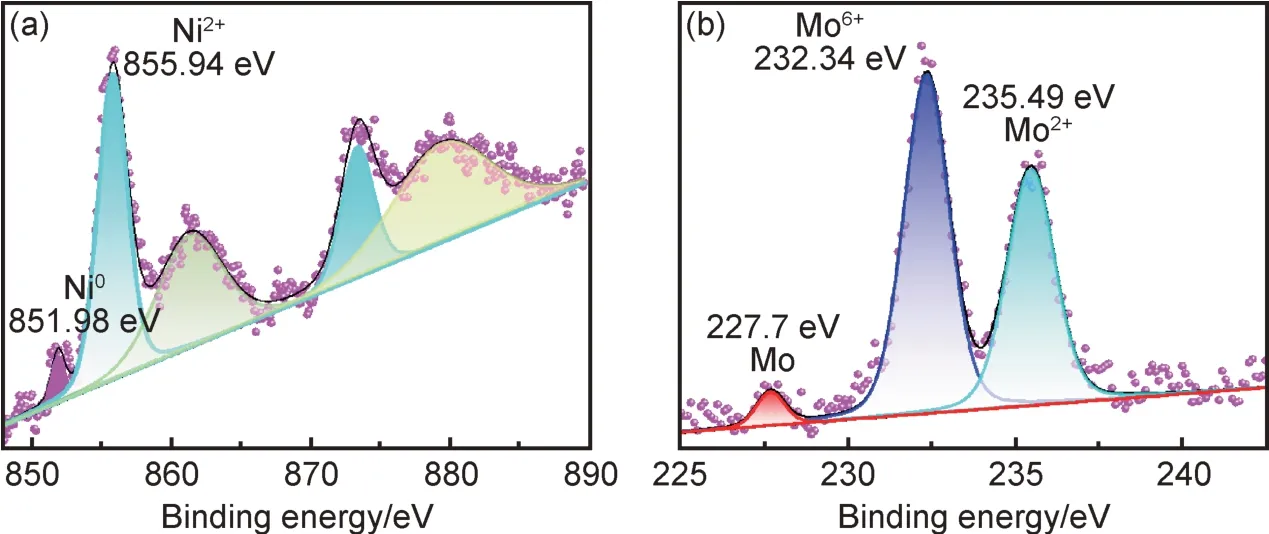
图5 Ni-Mo-1.4 合金镀层表面XPS 高分辨谱图 (a)Ni2p;(b)Mo3dFig.5 XPS high-resolution spectra of Ni-Mo-1.4 alloy coatings (a)Ni2p;(b)Mo3d
综上,初步判定Ni-Mo 合金镀层的沉积动力学为:在低的极化电位时(≤-1.0 V),合金镀液主导镍络合离子的还原,钼酸盐中间价态产物(MoOx)形成相对困难,电极表明呈现多孔的纳米镍镀层,表现为阻抗谱高频段压扁的容抗弧(图2(a),(b)),电极反应式如式(1)和式(2);当极化电位进一步增加时,电极附近的[Mo7O24(C6H7O7)]5-络合离子开始发生还原反应,生成中间价态的MoOx,造成合金镀层中的Mo 和O 含量显著增加(图3(c-3))。根据文献[11-12],钼酸盐还原的中间产物主要为MoO2,且电极附近的Ni2+将进一步促进MoO2转化成镍钼氧化物(MoO2Ni4),电化学反应式见式(3)和式(4)。此外,合金镀液中柠檬酸因配位数发生变化而离解出的H+在该极化区间容易被还原成H 原子(反应式(5)),并聚集形成H2通过电极表面的中间吸附体间隙不断逸出,但随着极化电位进一步增加,中间吸附体厚度明显增加,H2逸出能力下降,大量氢原子滞留在中间吸附体内部,因此在H 原子作用下,中间吸附体会被还原成Ni-Mo合金,而根据图3(d-3),(e-3)能谱分析可知,最终合金镀层中Ni/Mo 化学计量比约为4,因此推断电极表面发生的反应式如式(6)所示。需要指出,尽管在高极化电位下Ni-Mo 合金镀层接近Ni4Mo 组成,但是镀层的晶化程度较低,仍然以非晶形态存在(图4)。
通常,沉积过程中镀层的内应力(拉应力)常因镀层厚度增加而增大,而内应力不仅影响镀层界面结合力,同时诱导镀层内部出现微裂纹,从而影响镀层催化稳定性和催化活性。在电镀过程中,镀层厚度与沉积速率成正比,而沉积速率受界面电荷转移速度和镀液中离子扩散速度共同控制,不同极化电位下电极上电荷转移速度和溶液中离子扩散速度不一致,造成界面电化学反应速率不同,因而沉积速率也不同。图6为Ni-Mo 合金镀层在不同极化电位下的沉积速率。可以看出,镀层的沉积速率随着极化电位增加而增大,说明极化电位增大,电极表面的电荷转移加快,沉积速率增加,但当极化电位增加至一定值后(>-1.4 V),沉积速率减缓,说明此时镀液中离子扩散速度是速度控制步骤,即镀液中离子扩散速度决定镀层的沉积速率。另外,由于在大的极化电位下,镀层沉积速率较大,厚度显著增加会导致拉应力快速上升,造成镀层产生裂纹,如图3 所示。
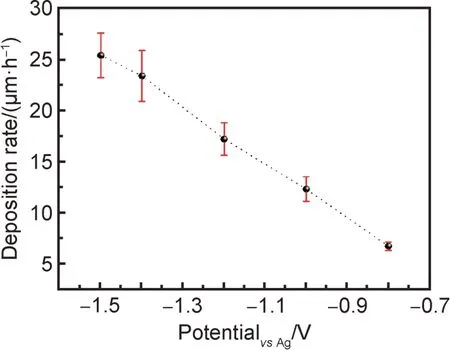
图6 Ni-Mo 合金镀层在不同极化电位下的沉积速率Fig.6 Deposition rate of Ni-Mo alloy coatings at different polarization potentials
2.2 Ni-Mo 合金镀层的析氢催化性能
不同极化电位制备的Ni-Mo 合金镀层不仅成分存在差异,其微观形貌也明显不同,两者均影响Ni-Mo 合金镀层电催化析氢性能。图7 为Ni-Mo 合金镀层析氢催化性能。从图7(a)的 LSV(linear sweep voltammetry)曲线可知,Ni-Mo 合金镀层析氢过电位随极化电位增加先减小后增加,即Ni-Mo-1.4合金镀层过电位最小,继续增加极化电位,其析氢过电位又显著增加。Tafel 斜率是衡量电极析氢动力学的重要参数之一,可根据Tafel 斜率大小判断析氢反应速度控制步骤,如Volmer步骤主要以电极吸附氢为速度控制步骤(H2O+e+*→H*+OH-,120 mV·dec-1),Heyrovsky 步骤主要以电化学脱附为速度控制步骤(H*+H2O+e →H2+OH-+*,40 mV·dec-1),Tafel步骤主要以化学脱附为速度控制步骤(2H*→H2+*,30 mV·dec-1)[17]。图7(b)为Ni-Mo 合金镀层的Tafel斜率,可以看出,Ni-Mo-1.4 合金镀层具有最小的Tafel 斜率(48.7 mV·dec-1),说明合金镀层上的析氢反应速度主要受Heyrovsky-Heyrovsky 混合控制。图7(c)给出了Ni-Mo 合金镀层在10 mA·cm-2和100 mA·cm-2电流密度下的析氢过电位,可明显看出Ni-Mo-1.4 合金镀层的析氢过电位较小(η10=51 mV,η100=149 mV),说明其具有良好的电极反应动力学。图7(d)为Ni-Mo-1.4 合金镀层电催化析氢稳定性测试,可以看出,合金循环析氢1000 周次后其电位下降幅度较小(Δη100=11 mV),而且经持续催化后其表面形貌没有发生明显变化(图7(d)插图),仅微裂纹数量较初始状态有所增加,说明持续的H2逸出会加剧镀层内部微裂纹的扩展,同时引起表面少许颗粒剥落。由于镀层属于层状堆积结构,表面微小脱落不会造成Ni-Mo 合金镀层催化性能的大幅下降,因此利用深共熔体系电沉积制备的Ni-Mo 合金镀层不仅具有良好的催化活性,同时有着较好的催化稳定性,这些都得益于深共熔溶剂对合金成分的有效调控,并极大地提高镀层的界面结合力。表1 总结了已有报道的Ni-Mo双金属或Ni-Mo-X三元合金析氢催化剂的过电位及Tafel 斜率,通过数据分析,可知氯化胆碱-乙二醇深共熔体系中电沉积制备Ni-Mo/Cu 合金镀层具有更加优异的电催化析氢性能[18-25]。

表1 Ni-Mo/Cu 与其他镍基催化剂电催化析氢中的过电位及Tafel 斜率Table 1 Overpotentials and Tafel slopes between Ni-Mo/Cu and other reported Ni-based catalysts for HER
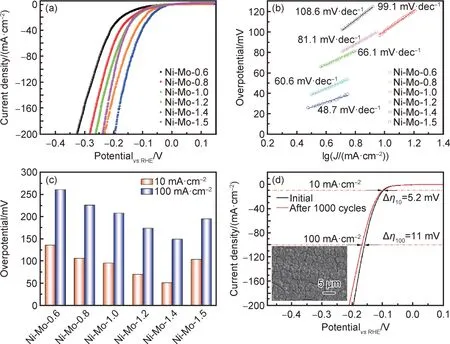
图7 Ni-Mo 合金镀层的析氢催化性能(a)LSV;(b)Tafel 斜率;(c)过电位;(d)Ni-Mo-1.4 合金镀层析氢稳定性Fig.7 HER catalytic performance of Ni-Mo alloy coatings(a)LSV;(b)Tafel slope;(c)overpotential;(d)catalytic stability of Ni-Mo-1.4 alloy coatings
电化学活性面积(electrochemical active surface area,ECSA)也是评价催化剂性能好坏的重要参数之一,通常与电极/溶液界面的双电层电容(Cdl)成正比[26],即Cdl越大,ECSA 越大,催化剂活性位点越多,催化性能越好。图8 为Ni-Mo 合金镀层不同扫描速率下循环伏安曲线及电流密度-扫描速率线性拟合图。可以看出,扫描速率越大,Ni-Mo 合金镀层界面双电层电容充放电电流越大;相同扫描速率下,极化电位越大,双电层电容充放电电流也越大(图8(a)~(e)),但极化电位增大至一定值后,电极界面双电层电容充放电电流开始减小(图8(f))。图8(g)给出了不同极化电位下Ni-Mo 合金镀层界面的双电层电容,可以看出,Ni-Mo-1.4 合金镀层的电容最大(Cdl=22.1 mF·cm-2),说明Ni-Mo-1.4 合金镀层的电化学活性面积最大,在此合金表面可以产生更多的活性位点,因此表现出优异的析氢催化活性。

图8 Ni-Mo 合金镀层的电化学活性表征 (a)~(f)Ni-Mo-0.6,Ni-Mo-0.8,Ni-Mo-1.0,Ni-Mo-1.2,Ni-Mo-1.4 和Ni-Mo-1.5 合金镀层不同扫描速率CV 曲线;(g)双电层电容;(h)本征交换电流密度Fig.8 Characterization of electrochemical activity for Ni-Mo alloy coatings (a)-(f)CV curves at different scan rates of Ni-Mo-0.6,Ni-Mo-0.8,Ni-Mo-1.0,Ni-Mo-1.2,Ni-Mo-1.4 and Ni-Mo-1.5 alloy coatings;(g)double layer capacitance;(h)intrinsic exchange current density
同理,本征交换电流密度(j0,real)也是衡量电催化剂本征催化活性的一个关键指标,而成分是影响其本征活性的重要因素。为进一步理解极化电位对Ni-Mo 合金镀层本征活性的影响规律,对其本征交换电流进行考察,如图8(h)所示。本征交换电流密度(j0,real)是表观交换电流密度(j0,app)与实际工作电极面积(Sreal)的比值(j0,real=j0,app/Sreal),其中Sreal=Cdl/k(k为单位面积金属汞的双电层电容,通常为20 μF·cm-2),j0,app=10-a/b(a为Tafel 拟合线的截距,b为Tafel 斜率)[27]。可以看出,Ni-Mo 合金镀层的本征活性与合金镀层中Mo 含量成正比,Mo 含量越高,其本征活性越高(Mo=28.6%,j0,real=1.07×10-3mA·cm-2)。值得说明,虽然Ni-Mo-1.5 合金镀层本征活性较高,但其Tafel 斜率以及过电位较大(图7(b),(c)),可能因为其组成颗粒粗大,表面粗糙度明显降低,能够暴露在外的电化学活性面积较小(Cdl=5.21 mF·cm-2),表观活性相对较小,综合催化性能下降。
3 结论
(1)随着极化电位增加,Ni-Mo 合金镀层的成分经历Ni →Ni+MoO2(MoO2Ni4)→Ni4Mo 演变。
(2)随着极化电位增加,Ni-Mo 合金镀层析氢过电位先减小后增大,其中Ni-Mo-1.4 合金镀层析氢过电位最小(η10=51 mV),且具有较小的Tafel 斜率(48.7 mV·dec-1)和较大的电化学活性面积(Cdl=22.1 mF·cm-2),表明其具有良好的析氢催化活性。
(3)Ni-Mo-1.4 合金镀层表现出良好的催化稳定性,循环析氢1000 周次后电位下降较小(Δη100=11mV),说明深共熔溶剂有助于提升镀层与基底的界面结合力。
