误诊为CIDP的神经元核内包涵体病1例报告
2023-12-18赖有连黄志珍罗聪丽
赖有连, 黄志珍, 罗聪丽, 林 攀
神经元核内包涵体病(neuronal intranuclear inclusion disease,NIID)是一种罕见的、进展缓慢的神经系统退行性疾病,临床常表现肢体运动感觉异常、意识障碍、癫痫样发作、智能减退、共济失调、周围神经病变、自主神经功能障碍等,常误诊为其他神经系统疾病,其病理学特征是在中枢神经系统、周围神经系统以及内脏器官的细胞中存在嗜酸性玻璃样核内包涵体[1]。既往诊断多依赖于尸检。随着日本学者Sone 等人[2]提出的可由皮肤活检确诊此病,本病报道数量增加。经过福建省龙岩市第二医院伦理委员会的审查同意,现将通过皮肤活检病理和基因检测确诊的1例NIID患者的临床诊疗经过进行报道。
1 病例资料
患者,男,46 岁,以“反复双下肢无力8年,再发1月余”为主诉于2017年10月9日入院。8年前开始出现双下肢无力,表现为双下肢站立、步态不稳,伴有全头部胀痛,出现反复恶心呕吐,伴有勃起功能障碍,曾反复出现一过性意识不清,就诊于外院完善腰穿检查后考虑慢性炎性脱髓鞘性多发性神经根神经病(chronic inflammatory demyelinating polyradiculoneuropathy,CIDP)[3],治疗后症状有好转(具体治疗不详),但上述症状仍反复再发并逐渐加重。既往素健,否认药物滥用史、中毒史及家族性遗传病史。查体:体温36.9 ℃,脉搏80 次/min,呼吸20 次/min;血压130/90 mmHg,心肺腹查体未及明显异常。专科检查:神志清楚、对答切题,计算力及定向力正常,双侧瞳孔等圆等大、对光反射灵敏,眼球运动正常、未见水平及垂直眼震,额纹对称、双侧鼻唇沟对称,伸舌居中、悬雍垂居中、双侧咽反射弱,四肢肌张力减低,双上肢肌力5 级、双下肢肌力4 级,四肢腱反射弱,四肢末端肌肉萎缩,深浅感觉双侧对称正常,双侧病理征阴性,脑膜刺激征阴性,共济试验正常。蒙特利尔认知评估量表(Montreal Cognitive Assessment,MoCA):22分。辅助检查:血常规、肝肾功能电解质、乙肝二对半、肌酸激酶及同工酶、凝血功能、血脂、甲状腺功能、血氨、血乳酸、梅毒螺旋体抗体测定、人类免疫缺陷病毒抗体、免疫结缔组织病全套、血管炎全套、肿瘤标志物全套等常规实验室检查均未见异常。入院诊断:双下肢无力待查,CIDP可能。肌电图提示:左侧正中神经末梢潜伏期稍延长4.4 ms,双侧正中神经运动传导速度均减慢;双侧尺神经末端潜伏期延长(左侧3.9 ms,右侧4.0 ms),复合肌肉动作电位(compound muscle action potential,CMAP)波幅下降;双侧腓总神经CMAP波幅明显下降、运动传导速度明显减慢;双侧胫神经F波出波率下降、最短潜伏期延长;双侧胫神经H反射未引出。肌电图结果详见表1。脱髓鞘灶。因受限于当时对NIID 的认知不足,未进一步诊断,治疗上予营养神经、改善微循环等处理,症状有改善,建议上级医院进一步诊治。

表1 肌电图结果
2020年9月7日患者因双下肢无力、反复头痛、恶心呕吐再次就诊我院,完善头部弥散加权成像(diffusion weighted imaging,DWI)(见图1)可见双侧大脑皮髓质交界区呈弥散受限高信号病灶,考虑NIID可能,取得患者及其家属同意,完善皮肤活检病理检查和NLCGGC相关疾病谱基因检测,遂于左侧踝关节上10 cm 处取皮肤活检送首都医科大学附属北京天坛医院行病理检查,免疫组织化学染色结果提示:部分汗腺细胞、脂肪细胞和纤维细胞的细胞核内可见P62、泛素抗体强阳性染色的包涵体(见图2)。诊断NIID明确。
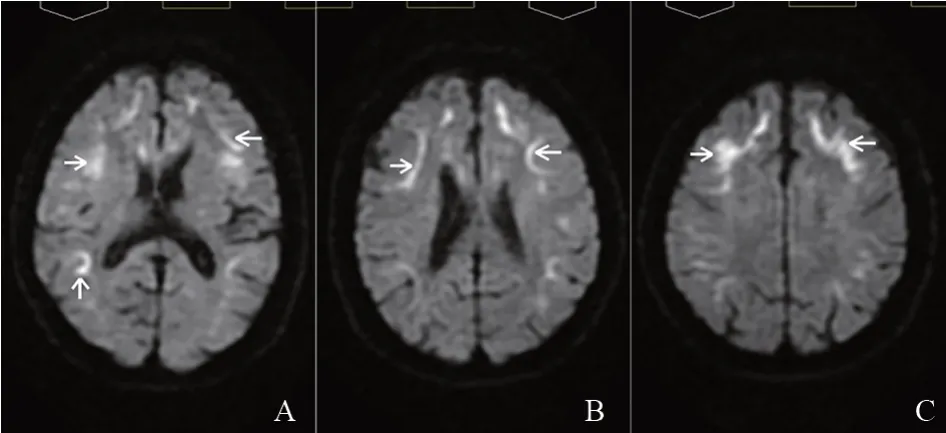
图1 头部核磁共振图像
腰穿脑脊液检查:压力100 mmH2O;常规、生化、免疫、三大染色及细胞学结果均未见明显异常。外院查头部磁共振平扫示双侧额颞顶枕皮质下对称性该患者基因检测结果(见图3)提示NOTCH2 NLC基因中GGC异常扩增次数为142次。
2 讨 论
NIID 是一种罕见的慢性进展性神经变性疾病,根据发病的年龄可分为婴儿型、青少年型和成人型,Sone等[1]据家族发病情况将NIID分为家族型及散发型,又据早期临床表现分为痴呆型和肢体无力型。典型成人型NIID 包括认知障碍和白质脑病,临床表现无明显特异性。一般来说,散发型NIID 发病年龄较大,家族型NIID 发病年龄较小,且肢体无力为首发症状较多[4]。杨帆等人[5]将NIID 主要症状归纳为3 个类型:(1)中枢神经系统受累,出现痴呆、共济失调、发作性意识障碍、行为异常、亚急性脑炎样表现、强直、震颤、癫痫发作、卒中样发作等;(2)周围神经受累,出现感觉障碍、远端肌力下降;(3)自主神经受累,表现为瞳孔缩小、尿失禁、呕吐、晕厥等。本例患者以反复进行性双下肢无力症状起病,病程进展过程中出现周围神经损害、性功能障碍、反复发作性意识障碍、轻度认知功能障碍等表现,且家属无相关症状,符合散发型NIID 表现。但该患者症状进展超过8周,呈慢性进展病程,表现为对称性双下肢无力,四肢腱反射减弱,外院诊断为CIDP,我院肌电图提示周围神经传导速度减慢、传导阻滞,无明显中毒、遗传、肿瘤、自身免疫疾病等其他周围神经病的证据,也支持CIDP 的肌电图表现[6],导致该病例极易误诊。因此,NIID的临床表现具有很强的异质性。
NIID的特征性影像学表现为DWI上皮髓质交界处(U型纤维)出现有高信号,这种征象随着疾病进展,病灶将不断地沿皮质向后延伸,不会向髓质深部延伸,陈为安等人[7]将其命名为“皮质下绸带征”。该患者的典型DWI特征为我们确诊NIID提供了关键线索。
进一步确诊NIID 主要依靠病理活检,在2011年Sone 等人[8]报道了在皮肤活检的标本中发现了脂肪细胞、成纤维细胞和汗腺细胞内可见嗜酸性核内包涵体,这些核内包涵体泛素化阳性。2019年,唐北沙教授团队[9]通过长读长测序(longreadse-quencing,LRS)首次揭示了NIID 致病机制与NOTCH2NLC基因中GGC异常重复扩增相关,且NIID临床表现的多样性可能与NOTCH2NLC基因5'区域GGC病理性重复次数相关,一般认为GGC重复扩增次数超过60次具有致病性。
综上所述,NIID临床表现多样,其病理表现为神经元核内出现嗜酸性包涵体,影像学可表现为经典“绸带征”,症状上可与多种神经变性病相叠加,其发病与NOTCH2NLC基因5'区域GGC 病理性重复次数相关。本例NIID 如无影像学检查作为线索,将一直被认为是CIDP,这提示我们在临床诊疗中发现周围神经病变的患者,也应当重视和排查中枢神经系统是否累及。因此,呼吁我国NIID 协作组开展针对NIID的研究,深入了解其发病机制,并不断完善制定相关诊断标准和流程。
伦理学声明:本研究方案经由福建省龙岩市第二医院伦理委员会审批(批号:LYEYEC 2022-005),患者已签署知情同意书。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:赖有连、林攀负责论文设计;赖有连负责绘制图表和撰写论文;赖有连、罗聪丽、黄志珍、林攀负责数据及文献收集;林攀负责拟定写作思路、指导撰写文章并最后定稿。
