电化学阴极制氢耦合阳极废水污染物降解的研究进展
2023-12-18冯江涛孙娅星林长征朱金薇延卫
冯江涛, 孙娅星, 林长征, 朱金薇, 延卫
(1. 西安交通大学能源与动力工程学院, 710049, 西安; 2. 陕西电器研究所, 710025, 西安)
随着全球环境状况的不断恶化与化石能源的日渐短缺,从传统化石燃料经济向绿色和可持续能源经济的转变至关重要。氢气具有能量密度高、用途广泛等优点,燃烧时仅产生水,且可以高效廉价地生产,被视为未来最具发展前景的可再生能源之一[1]。传统的氢气生产以化石燃料重整为主,成本较高,且在产氢的同时会生成大量的CO2,对环境造成污染[2]。电化学制氢是通过阴阳两极通电将水电解,同时生成氢气和氧气的过程。整个电解过程包括两个半反应[3],即阴极析氢反应(HER)
2H++2e-→ H2
(1)
和阳极析氧反应(OER)
H2O → (1/2)O2+2H++2e-
(2)
该方法产氢具有高效率、高纯度的优点,理论上可以实现零碳排放制氢,为国家“双碳”目标的实现做出贡献。运用电解水制氢,同时也可以将光能、风能或潮汐能生产的多余电力吸收转化为化学燃料,提高可再生能源的利用率[2]。在标准条件下,需要1.23 V的理论电势来驱动电化学水分解制氢,但由于阳极析氧反应四电子转移的缓慢动力学,需要较高的过电位才能实现水的电解反应,导致能量转换效率降低[4]。因此,需要开发能够高效催化阴极析氢反应和阳极析氧反应过程的催化剂(电极材料)。此外,阳极产生的O2不仅价值较低,而且在电解槽的长期运行中,即使使用膜进行分离,也可能与阴极产生的H2发生气体交叉,造成爆炸。H2、O2和电催化剂的存在也可能形成活性氧(ROS),使隔膜老化,降低电解槽的寿命[5]。
为解决以上问题,科研工作者提出一种将阴极还原产氢与阳极氧化降解污染物耦合的方法[6-7],以阳极氧化降解污染物取代动力学缓慢的OER来辅助制氢,在产氢的同时实现对污染物的降解。污染物应尽可能选取氧化电位低、还原性高、在氧化过程中产生有毒有害物质少、对电极影响小的物质。
目前,应用于耦合系统的污染物有醇类(氧化还原电位Eθ≈0.5~1.3 V,以饱和甘汞电极为参比)、酚类、醛类、肼(Eθ=-0.33 V)、尿素(Eθ=0.37 V)、胺、液氨、硫化物(Eθ=-0.48 V)等,与OER过程的理论电压1.23 V相比,具有一定的电位优势。根据不同的催化剂构建混合电解系统,比相应催化剂构建的水电解系统电池电压低0.1~0.4 V,污染物去除率/转化率可达90%以上,制氢法拉第效率接近100%,具体研究见后文。
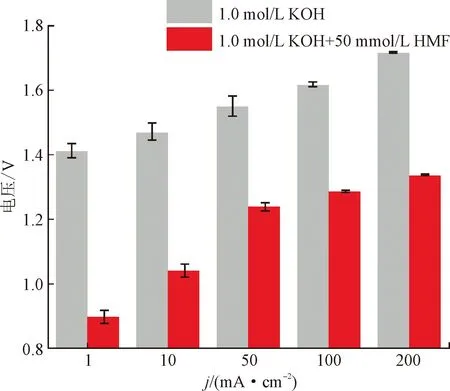
实际废水因排放场所、汇集管道等情况综合在一起,成分复杂,易受污染物浓度、pH、温度、污水流速、催化剂性质等诸多因素影响。污染物浓度不同,耦合体系所需要的耗能也不同,针对特定污染物制氢体系应有特定的浓度探究。pH是影响阳极降解污染物辅助制氢的另一重要因素。根据pH不同,可以构建不同性质的耦合体系,对相应的催化剂要求也更多。通常使用的电解液为1 mol/L KOH (pH =14)[7],为了更好地降低能耗,阴极使用0.5 mol/L H2SO4(pH=0)、阳极使用1 mol/L KOH (pH=14)的酸碱电解槽[8]也在蓬勃发展。虽然以阳极氧化污染物辅助制氢的策略极具发展前景,但这更多处于实验室阶段,工业应用方面尚未大规模成型。
实现高效制氢与治污耦合的关键之一是开发性能稳定的电极材料(催化剂)。催化剂表面氢原子的吸附和解吸应当处于平衡,这种催化剂活性通常使用氢吸附的自由能(ΔGH)来表示,以ΔGH接近0为最佳。金属催化剂的ΔGH与催化活性具有“火山”关系[9],即以金属催化剂的氢吸附键能(EM-H)为横坐标,交换电流密度的对数(lgj0)为纵坐标,催化剂在其中的位置分布形成“火山”,越靠近顶端析氢所需过电位越小,催化剂的性能越好,如图1所示。
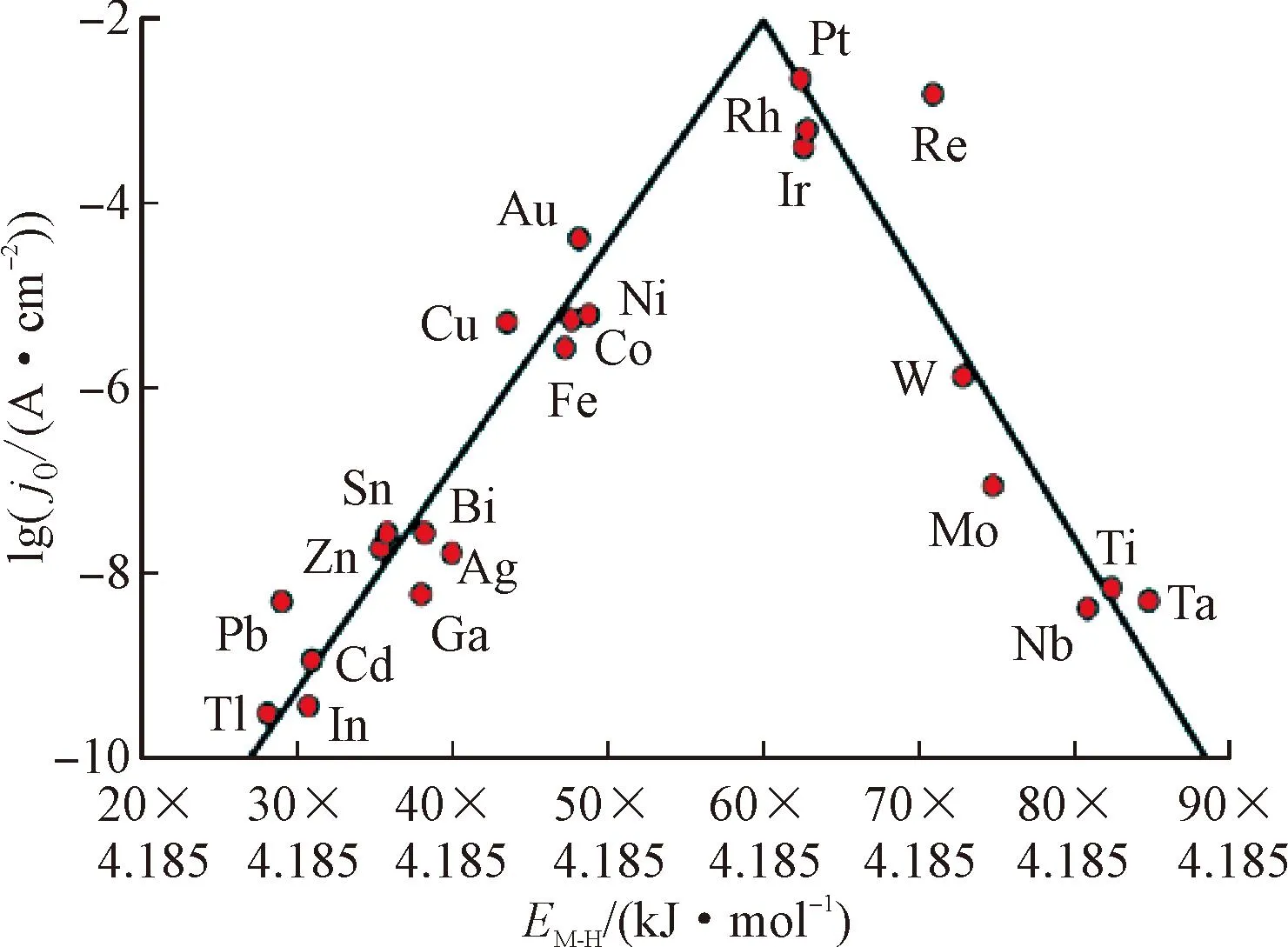
(a)酸性介质
贵金属(铂、铑、铱等)电极位于“火山”顶部,是HER的绝佳催化剂,具有最好的氢吸附能。Ouyang等[11]成功合成了RuO2纳米颗粒修饰的TiO2纳米阵列催化剂(RuO2@TiO2/TP),在酸性介质中,只需130、166 mV的过电位即可达到100、200 mA·cm-2的电流密度,在碱性和中性介质中同样具有出色的活性。Lin等[12]开发了一种负载在N掺杂石墨烯(NG)上的精细有序的Pt3Co/NPs催化剂(Pt3Co/NG-700),以13 mV的过电位实现了10 mA·cm-2的电流密度,展现了高HER活性,甚至优于Pt/C催化剂的28 mV过电位。
贵金属成本过高、资源稀缺,难以实现大规模应用。因此,以过渡金属催化剂为主的非贵金属基催化剂成为研究重点[13],镍[14]、钴[15]、铁[16]、钼[17]等过渡金属具有多种价态,可以通过杂原子掺杂、空位、异质结构、应变和相变等手段来调节电子构型,增强析氢性能[18]。Liu等[6]通过热处理合成了一种多元结构硫化物/磷化物催化剂(Ni3S2-Ni3P/NF),作为尿素-水整体电解的阳极和阴极,仅需1.65 V的电压即可达到100 mA·cm-2的电流密度,证明了利用非贵金属基催化剂进行制氢与治污的潜力,如图2所示。Zhang等[19]以沸石咪唑骨架(ZIF-67)为模板,通过溶解-再生、磷酸化制备了一系列CoP·xCoMoP NCs电极,当Co与Mo的原子比为 6∶5的最佳原子比时,制备的CoP·5CoMoP NC催化剂在酸性、碱性和中性条件下均表现出优异的活性和稳定性。
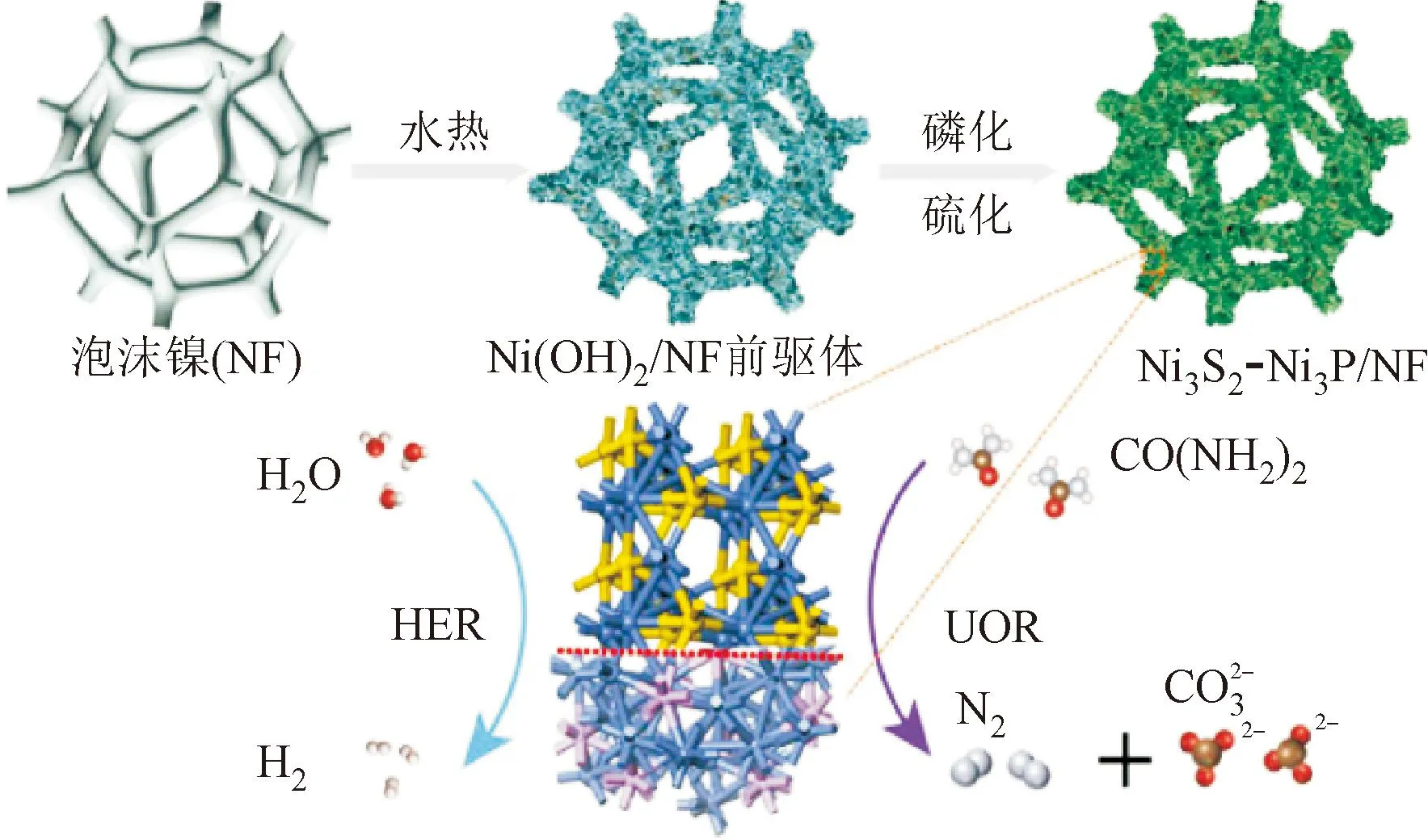
图2 尿素-水整体电解示意[6]
为对电催化剂的催化性能做出评估,基于活性、稳定性和效率等关键参数发展出以下评价指标[20-21]。
(1)过电位。在1.23 V的理论电压下,水电解在动力学上是可逆的,HER和OER反应很难发生,此时需要过量的电位来克服动力学势垒,即过电位(η)。通常,使用10 mA·cm-2的电流密度所对应的过电位值(η10)来比较不同催化剂之间的活性,该电流密度对应于太阳能转化为氢时约10%的效率,由电化学线性极化曲线(LSV)测试得出。
(2)塔菲尔斜率和交换电流密度。塔菲尔斜率是一个重要的动力学指标,可以通过塔菲尔方程计算得到,η=a+blogj。其中,a、b是塔菲尔常数,j是测量的电流密度。塔菲尔斜率取决于电荷转移系数和转移电子的数量,其值越小,表示当施加较高电压时,电流密度增加得更快,电催化反应动力学更佳。当η=0时,根据塔菲尔方程计算出的相应的j为交换电流密度(j0)。它反映了平衡条件下电子转移的固有速率,交换电流密度的幅度越大,电荷传输速率越大,催化剂对水电解的活性越大。
(3)稳定性。稳定性用于评估催化剂在长期运行期间保持活性的能力,是评价电催化剂是否具有实际应用潜力的重要参数。通常,通过在固定电位下进行CV循环并监测循环过程中的过电位变化判断,或在恒电流或恒电位下进行电解测量。只要过电势增加不超过30 mV并且稳定性测试后的总活性降低不超过5%,催化剂就可以被认为具有良好的稳定性。
(4)法拉第效率。在电解过程中有许多副反应发生,生成臭氧、过氧化氢和超氧化氢等,造成电力损失。此外,催化位点还必须经历连续的氧化和还原循环。因此,电解过程中的整体能量损失是不可避免的。法拉第效率用于描述由外部电路提供的促进电化学反应的电子转移效率,定义是实验检测的H2或O2的物质的量与理论计算的H2或O2的物质的量之比,用来排除其他反应的干扰。法拉第效率可以通过使用水-气置换法或气相色谱法来分析气体产量并计算得出。
此外,阳极催化剂降解污染物过程中,还有电化学阻抗谱(EIS)、电化学粗糙度(Rf)、污染物降解率(ηc)、化学需氧量(COD)去除率(ηCOD)等参数[22-23]。
(1)电化学阻抗谱。简单来说,EIS将电化学过程模拟为一个电路模型,用于评估电极和溶液界面处的电子转移。催化剂阻抗值越低,意味着该催化剂界面电子转移能力越强。
(2)电化学粗糙度。电极每个表观几何面相加的实际表面积。
(3)污染物降解率。ηc=(C0-Ct)/C0×100%。式中:C0为污染物初始浓度;Ct为t时刻污染物浓度。
(4)COD去除率。ηCOD=(M0-Mt)/M0×100%。式中:M0为初始COD;Mt为t时刻COD。
除催化剂外,电解槽的设计也十分重要。目前,电解水制氢所用电解槽主要有3种:碱性水电解槽(AWE),质子交换膜电解槽(PEM),固体氧化物电解池(SOEC)[24]。AWE使用碱液作为电解质,无需贵金属作催化剂,在3种技术中商业化程度最高[25],但存在电极腐蚀和隔膜损耗的问题。PEM电解槽使用聚合物膜为固体电解质,允许质子通过并阻碍气体传输,制氢率和所得氢气纯度都较高,但具有成本高、寿命短和对环境影响较大的缺点[26]。SOEC需要在500~1 000℃的高温下反应,通常以固体陶瓷作为电解质,并可与太阳能、风能等间歇性能源相连实现高效制氢,但SOEC目前尚未投入实际应用[27]。由于与制氢耦合的污染物降解反应大多在碱液中更为有利,所以混合水电解槽以AWE居多。值得注意的是,混合水电解减少了O2的析出,阳极产物多为液相,在设计方面可以实现无膜装置,降低成本并简化操作。另外,HER过程在酸性介质中的动力学比在碱性介质中更快速,因此可以设计阴极酸性、阳极碱性的不对称电解槽来更好地实现低能耗制氢[8, 28]。
为实现电化学制氢与治污的耦合,已有很多学者进行了尝试。一般而言,溶于电解液且理论氧化还原电位小于水的物质即可作为辅助制氢的电解质。本文系统总结了目前发展中的可以用于耦合制氢的污染物,其中一些最终矿化为水,另一些甚至可以生成附加化学品,提高电化学反应的电子利用率。
1 羟基化合物氧化反应与HER耦合
1.1 醇类氧化反应与HER耦合
醇类氧化反应(AOR)因应用于直接醇燃料电池(DAFC)和精细化工合成工艺而成为实现可持续能源生产的重要方法之一[29]。甲醇、乙醇、丙醇、甘油及生物质衍生醇等的电催化氧化为氢气生产过程提供了更大的电流密度,降低了整个电解池的电压。伯醇中的大多数在0.5~1.3 V之间的阳极电位(以饱和甘汞电极为参比)下氧化,醇类可以完全矿化为水和CO2,更可以电解产生增值产品[30]。表1列出了不同电催化剂对于醇类氧化的活性。
表1 用于醇类氧化反应与HER耦合的电催化剂及其活性
虽然醇类氧化反应在降低能耗方面极具优势,但其产物多样,对易于分离的产物进行选择性反应是研究的重点。迄今为止,贵金属(铂、钯、金等)及其合金仍然是醇类的催化氧化中最有效的催化剂。Zhou等[35]开发了一种通过自发电流还原合成的单分散Ru锚定多孔PtNi合金(Ru1-Pt3Ni/NF)作为双功能电催化剂,实现了乙醇向乙酸盐的高选择性电氧化耦合制氢。
TEM图像显示,Ru掺杂的Pt3Ni形成均匀的多孔结构(见图3(a)),这大大增加了催化剂的比表面积,为乙醇的电催化氧化提供了更多的反应位点。采用Pt3Ni/NF作为HER催化剂、Ru1-Pt3Ni/NF作为阳极催化剂,以2 mol/L KOH和2 mol/L C2H5OH作电解质,构建双功能电解槽。该体系仅需0.7 V电解电压即可达到125 mA·cm-2的电流密度并用于阴极H2生成(见图3(b)),单次测试(时长0.5 h)中,制氢法拉第效率达到94%。

(a)TEM图像
由于贵金属成本高、储量少,所以开发具有高化学活性的低成本电催化剂也成为研究重点,并取得了一定的进展。特别是一些过渡金属基催化剂,甚至可以达到与贵金属相当的活性[36]。Hao等[14]通过在泡沫镍基底上制备Ni(OH)2形成纳米片层状的Ni(OH)2/NF催化剂,该催化剂在1 mol/L的KOH溶液中,含有500 mmol/L甲醇为电解质时,只需1.52、1.62 V的槽电压就能分别驱动10、50 mA·cm-2的电流密度,而在不含甲醇的电解质中需要更高的槽电压(1.70、1.79 V),见图4(a)。实验结果表明:甲酸盐是阳极的唯一产物,阳极产物法拉第效率保持在几乎100%,见图4(b);阴极产氢的法拉第效率达92%以上,见图4(c)。密度泛函理论(DFT)计算表明,HCOOH是Ni(OH)2裸露(001)面的热力学优势产物,证实了甲醇转化为增值甲酸盐的高选择性。Mo具有多种价态和半填充d轨道,可以诱导电子再分布,优化催化剂和反应物之间的结合相互作用。Wang等[37]在碳布上构建了Mo掺杂的Co4N纳米片(Mo-Co4N)用于节能制氢和甲醇电氧化的耦合。使用Mo-Co4N作为HER与甲醇电氧化反应的双功能催化剂,可以以1.427 V的低电池电压驱动10 mA·cm-2的电流密度,比整体水分解系统低169 mV,甲醇转化为甲酸盐的法拉第效率高达90%以上。
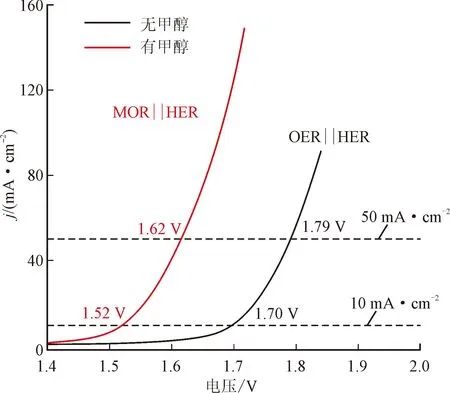
(a)LSV曲线
1.2 酚类氧化反应与HER耦合
酚类化合物毒性高,对人体、动植物等都有极大危害,电催化氧化是处理此类污染物的理想方法之一,具有无二次污染、降解效率高、反应条件温和等优点[38]。酚类氧化反应(POR)所需电压一般低于OER过程,与析氢半反应耦合可以一举实现酚类污染物降解和大大降低氢气生产的能耗两个目标。
2020年,Zheng等[39]用S2-取代多金属氧酸盐中的O2-制成S-NiP2Mo5催化剂,该催化剂展现出优异的HER活性、POR活性和稳定性。由于硫化之后催化剂的氢吸附自由能(ΔGH*)发生改变,降低了对H*的吸附,促进了H2的生成。经高效液相色谱(HPLC)技术对降解产物进行研究,苯酚降解是由于电催化作用而非吸附,并最终矿化为CO2和H2O。XPS分析表明,Ni2+先在电压下变为Ni3+,与苯酚发生反应后还原为Ni2+。以S-NiP2Mo5作为双功能电极构建电解槽,只需1.47 V电压就可获得25 mA·cm-2的电流密度,比整体水电解降低约0.4~0.45 V。随后,Qin等[40]制备了一种NiMoO4-NF电极,用于苯酚氧化辅助制氢。该电极表面大量垂直排列的纳米针有利于苯酚的扩散,促进了POR和HER的反应过程,HER过程中达到10 mA·cm-2的电流密度需要67 mV的过电位。同时,NiMoO4-NF对苯酚氧化的中间产物有较强的抗失活能力,其对苯酚的降解主要依靠表面羟基自由基的直接氧化过程。
传统的电化学氧化酚类所使用的阳极多为硼掺杂金刚石(BDD)电极[41]、金属氧化物电极[42-43]等,这些电极的活性和稳定性可能会因电极结垢而下降,具有低反应速率和效率。另外,化合物的浓度不同,降解速率也受传质过程的影响。酚类作为一种持久性有机污染物,因降解难度、适用电极的发展等缘故,与HER耦合体系的工作相对较少,具有深入研究的可能。
2 醛基化合物氧化反应与HER耦合
电催化氧化醛是合成有机酸的重要途径之一,同时也是辅助低压氢气生产的良好反应。在醛类氧化反应中,生物质衍生分子5-羟甲基糠醛(HMF)和糠醛(FUR)等的氧化是研究重点。HMF是C6碳水化合物的脱水产物,作为一种中间产物,可以被氧化生成2,5-二甲酰呋喃(DFF)、5-羟甲基-2-呋喃甲酸(HMFCA)、5-甲酰基-2-呋喃甲酸(FFCA)和2,5-呋喃二羧酸(FDCA),用以生产绿色聚合物、药品、树脂、溶剂、杀菌剂等。其中,2,5-呋喃二羧酸(FDCA)有望取代对苯二甲酸用来生成高性能聚合物,同时也是呋喃酸聚乙烯(PEF)的原料之一[44-45]。表2列出了HMF氧化反应与HER耦合的最新研究工作进展。

表2 用于HMF氧化反应与HER耦合的电催化剂及其活性
HMF羟基和醛基的氧化可分为两种反应途径。第一种首先氧化羟基从而产生2,5-二甲酰基呋喃(DFF),第二种通过醛基氧化生成5-羟甲基-2-呋喃甲酸(HMFCA),见图5。然后,DFF和HFCA被氧化为5-甲酰基-2-呋喃甲酸(FFCA),并进一步产生2,5-呋喃二羧酸(FDCA)[50, 52]。在强碱性介质(pH ≥13)条件下,HMF通过氧化为HMFCA后生成FDCA是主要反应路线,这更加有利于HMF的氧化与强碱环境下电解制氢的耦合,实现氢气与FDCA的共生产。代表性实验有Sun团队[46]通过研究直接磷化在泡沫镍上制备3D Ni2P NPA/NF电极的电催化性能,证实HMF的氧化反应比OER耗能少,更易实现。通过HPLC监测HMF及其氧化产物在电解过程中的浓度变化,得出本实验可能遵循由HMFCA到FDCA的路线,H2和FDCA生产的法拉第效率分别可达100%和98%。与全电解水相比,HMF氧化辅助制氢可以使H2的生产电压降低至少200 mV。
图5 5-羟甲基糠醛电化学氧化的两种途径
在HMF的氧化中,贵金属基电催化剂具有低起始电位的优点,但它们仅提供极低的电流密度。同时,以Ni基材料为代表的非贵金属催化剂表现出极高的活性,然而Ni对于H2的强吸附限制了其对HER的催化活性。
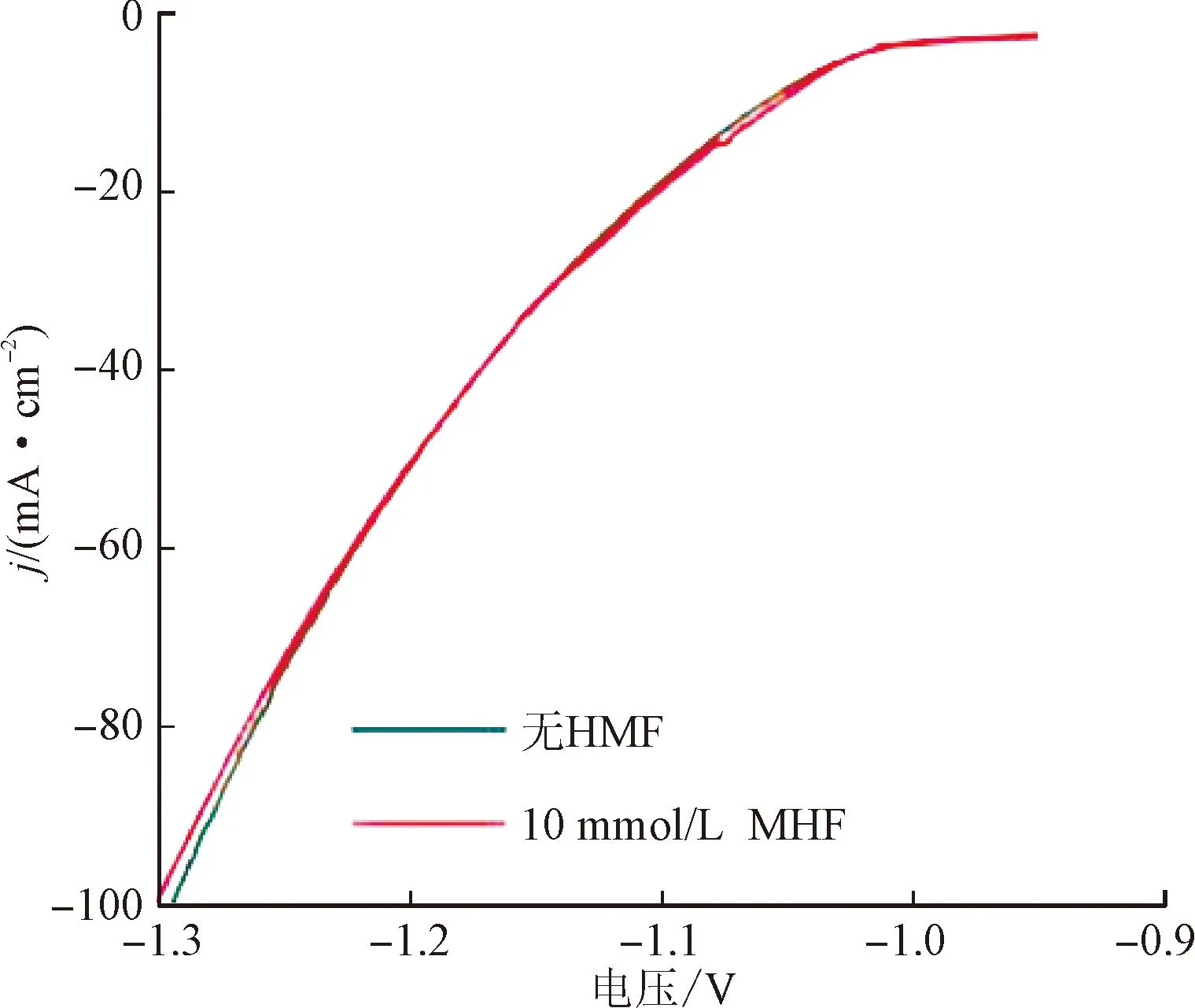
Liang等[50]使用Ni3N-V2O3作催化剂,利用镍和钒氧化物之间的界面效应减少了对H2的强吸附,完成了FDCA和H2的高效生产。Ni3N-V2O3对HMF的氧化仅需230 mV的过电位就可以达到10 mA·cm-2的电流密度,比OER过程降低了140 mV的过电位。HMF的存在对HER过程基本无影响(见图6(a),图中横坐标电压以可逆氢电极RHE作参比),表明该催化剂对HMF有很强的耐受性。HPLC分析(见图6(b)~(c))表明,HMF的氧化主要沿第二种路径进行,FDCA的产率达96.1%。
(a)LSV曲线
除Ni基催化剂外,含Cu催化剂因对OER几乎无活性,且能以各种电子状态存在,成为HMF电催化氧化的高效催化剂之一。Zheng等[48]研究了一种铜掺杂镍纳米管(NiCu NTs)催化剂,在HER与HMF氧化耦合的电解槽中驱动100 mA·cm-2所需的槽电压比传统水电解降低了350 mV,FDCA生产的法拉第效率达96.4%。
此外,还有Co基催化剂。Sun等[53]制备的CoxNiS@NF用于HER与HMF的耦合,明显降低了氧化电位(见图7(a),图中纵坐标电压以可逆氢电极RHE作参比)。至于双层氢氧化物(LDH)催化剂,Song等[49]开发了E-CoAl-LDH-NSA(见图7(b))等催化剂,对于HMF氧化和HER的耦合也具有较好的催化性能。
(a)CoxNiS@NF的氧化电位
HMF氧化辅助制氢有诸多好处的同时,也存在一些问题。首先,HMF的热稳定性和化学稳定性不甚完美,阻碍了其储存和工业化;其次,Ni、Co基等催化剂分别存在着起始电位过高、动力学过慢等缺陷;再次,目前实验室阶段仅添加以mmol/L计的HMF,无法实现工业规模。为使HMF氧化与HER耦合走向工业化,还需要进一步的探索研究。
3 含氮化合物氧化反应与HER耦合
3.1 肼氧化反应与HER耦合
肼是一种重要的工业原料,可作为喷气式发动机燃料、火箭燃料、抗氧剂、还原剂、高压锅炉给水脱氧剂等使用,在化工、制药、燃料电池等领域有着广泛的发展[54]。肼具有强毒性,属于2A类致癌物,对人体健康和环境方面都有很大危害。但是肼的电氧化电位很低,理论电压仅有-0.33 V[55]。
与醇、醛等物质的电化学氧化反应相比,肼的电化学氧化具有很多优势:①由于强还原性而使氧化电位更低;②氧化过程中不会生成温室气体或催化剂中毒物种;③具有快速氧化动力学,这使肼的氧化成为取代OER耦合制氢的极佳选择。表3为肼氧化反应(HzOR)与HER耦合的研究进展。

表3 用于肼氧化反应与HER耦合的电催化剂及其活性
HzOR与HER的耦合大多数都是在碱性电解质中进行的,对应的两个半反应[64]如下。阴极反应
2H2O+2e-→ H2+2OH-
(3)
阳极反应
N2H4+4OH-→ N2+4H2O+4e-
(4)
总反应
N2H4→ N2+2H2
(5)
Tang等[54]开发了基于Ni(OH)2/NF的Ni2P/NF纳米阵列催化剂,首次实现了HzOR辅助制氢的研究。该实验中,首先研究了Ni2P/NF的HzOR活性,证实以HzOR取代OER可以降低阳极反应的过电位。该催化剂在1 mol/L KOH和0.5mol/L N2H4的混合溶液中,分别仅需-290、18 mV的电压即可在HER和HzOR的过程中实现200 mA·cm-2的电流密度(见图8,图中横坐标电压以可逆氢电极RHE作参比),需要1.0 V的电池电压就能达到整体500 mA·cm-2的电流密度。
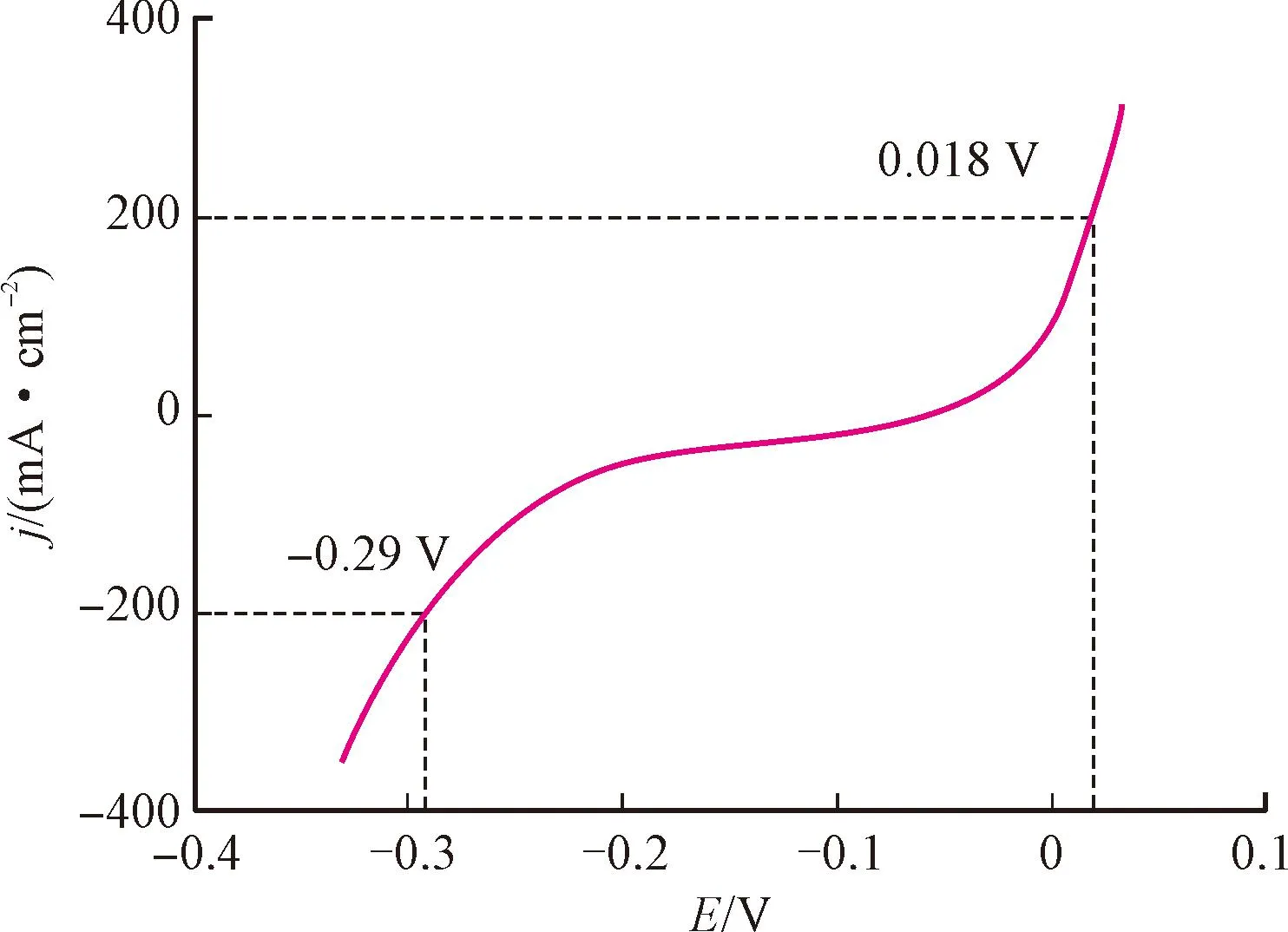
图8 Ni2P/NF电极HER和HzOR的LSV曲线[54]
此后,关于HzOR与HER耦合的研究逐渐增多。Co基材料因其优异的导电性和稳定性也被应用于该耦合电解系统。Zhang等[58]采用两步水热法,制成双功能管状CoSe2纳米片电极。该电极外表面由致密的纳米片相互连接而成,以暴露更多活性位点,便于传质和气体扩散。在加入了0.5 mol/L肼电解质的KOH溶液中,阴极CoSe2电极显示产氢法拉第效率高达98.3%(见图9(a)),对制氢过程中的电子利用效率非常高。图9(b)显示,组装好的HzOR与HER耦合的电解系统仅需 0.164 V 即可产生 10 mA·cm-2的电流密度,并可以在14 h内稳定制氢。Xu等[65]通过水热法结合低温磷化,成功制备了在钴泡沫上原位生长的CoxP@Co3O4纳米阵列(P-Co3O4/Co),电流密度为100 mA·cm-2时,水肼电解槽中的电池电压比只含1 mol/L KOH的电解槽低1.57 V。Hou等[62]采用CoS2中掺杂Mn原子的策略调节电子结构,在整体肼分解(OHzS)中分别需要111、329、447 mV即可驱动10、100、200 mA·cm-2的电流密度。Qian等[63]开发了在泡沫镍上原位生长具有丰富Ni3N-Co3N异质结构的分级多孔纳米片阵列(Ni3N-Co3N PNAs/NF),用于组装的OHzS系统中,只需要0.668、0.825、0.964、1.09 V的小过电位即可驱动100、200、300、400 mA·cm-2的电流密度。值得注意的是,为解决OHzS 系统的外部电源问题,在Qian等[63]的实验中,应用了直接肼燃料电池(DHzFC)的设计,见图9(c)。DHzFC以Ni3N-Co3N PNAs/NF为阳极,以负载在碳纸上的Pt/C电极为阴极。阳极含有1 mol/L KOH+0.5 mol/L N2H4的电解质,阴极含有1 mol/L KOH电解质,阴阳两极构成不对称电解质的电解槽,与OHzS形成自供电系统,实现自供电电解制氢。
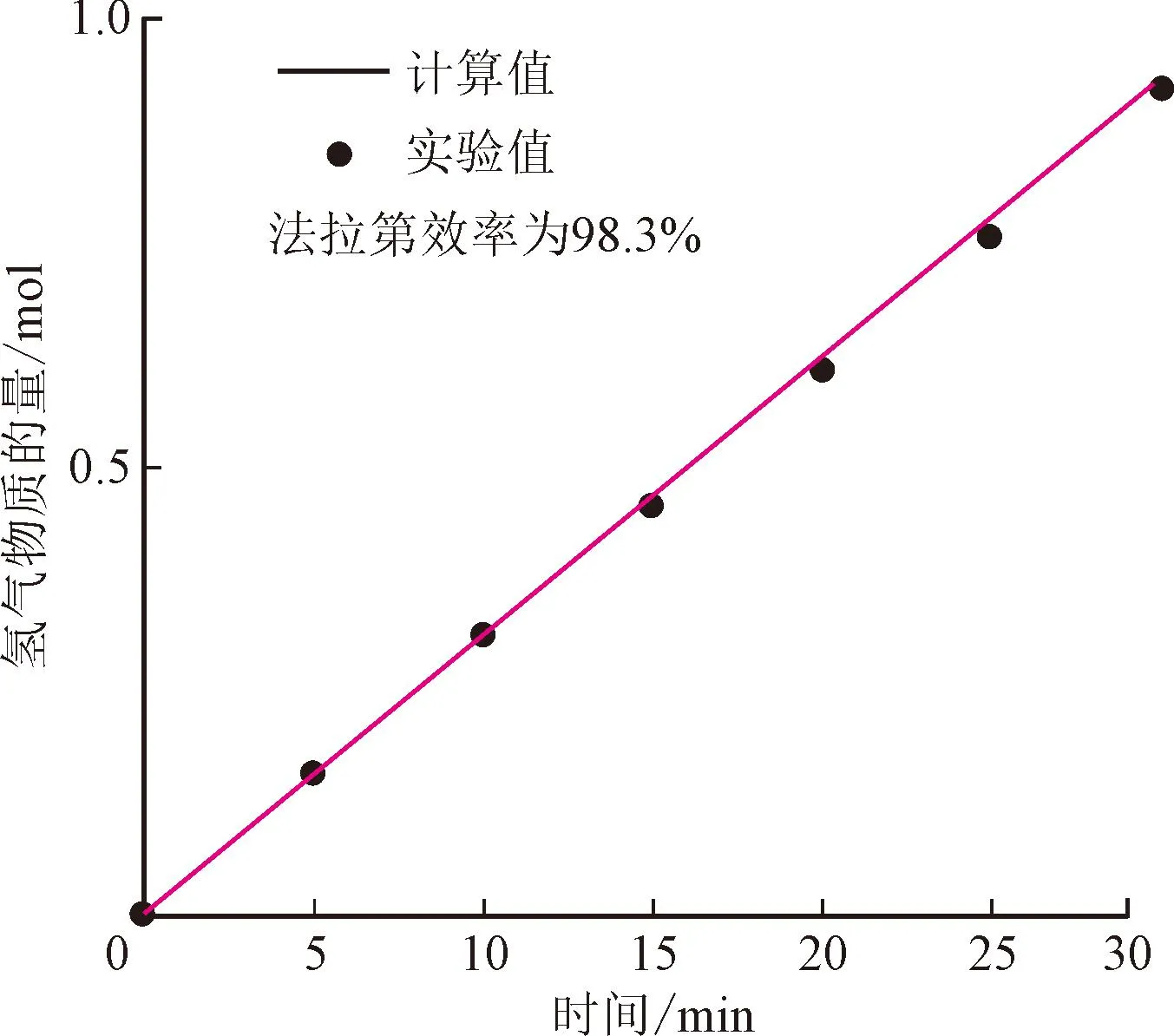
(a)制氢的法拉第效率
更早应用了DHzFC与OHzS自供电系统的还有刘熙俊所作的工作[66]与章根强教授课题组所做的工作[56]。前者以Fe掺杂的CoS2纳米片作为OHzS的双功能电极,DHzFC中则以Pt/C为阴极,将H2O2还原为H2O。以Fe-CoS2为阳极,将N2H4氧化为N2。该系统最大功率值在1.80 V达246 mW·cm-2,析氢速率为 9.95 mmol·h-1,法拉第效率为98%,表明该系统极具应用前景。后者将P、W共掺杂Co3N制备的PW-Co3N NWA/NF用于HzOR和HER的高效双功能电催化剂。电池电压为0.429 V时最大功率密度可达46.3 mW·cm-2,从肼到H2的总效率约为45.8%。对于大规模应用,肼的高毒性和不稳定性是最具挑战性的问题,所以其离实际应用还有大量的研究工作需要进行。
此外,最近还有研究针对HzOR和海水电解耦合制氢的研究报道。Sun等[67]以海水为阴极电解液,以1 mol/L KOH和0.5 mol/L肼为阳极电解液,中间由阴离子交换膜隔开组装成不对称电解槽。该系统可以在1.05 V的低电压下实现500 mA·cm-2的高电流密度,比直接电解海水制氢降低约45.6%的电压,并且无ClO-生成。这种通过将无成本的海水和工业上的肼污水来耦合制氢的系统,可以实现低成本、可持续制氢并完成肼污染物的降解。
3.2 尿素氧化反应与HER耦合
尿素废水主要来自尿素合成过程中的工艺排放、人或动物的分泌物以及尿素作为化肥的大量使用,这些未经处理的尿素废水可以自然转化为有毒氨释放到大气中,污染空气和饮用水,进而对人体健康产生影响[68]。电催化氧化技术被认为是降解尿素的有效方法,与此同时,尿素的毒性低、能量密度高、来源比较广泛,理论分解电位为0.37 V,远小于OER过程的其他化合物,尿素的电催化氧化(UOR)也被认为是极具前景的取代OER的反应。表4列出了目前UOR与HER耦合的一些工作。
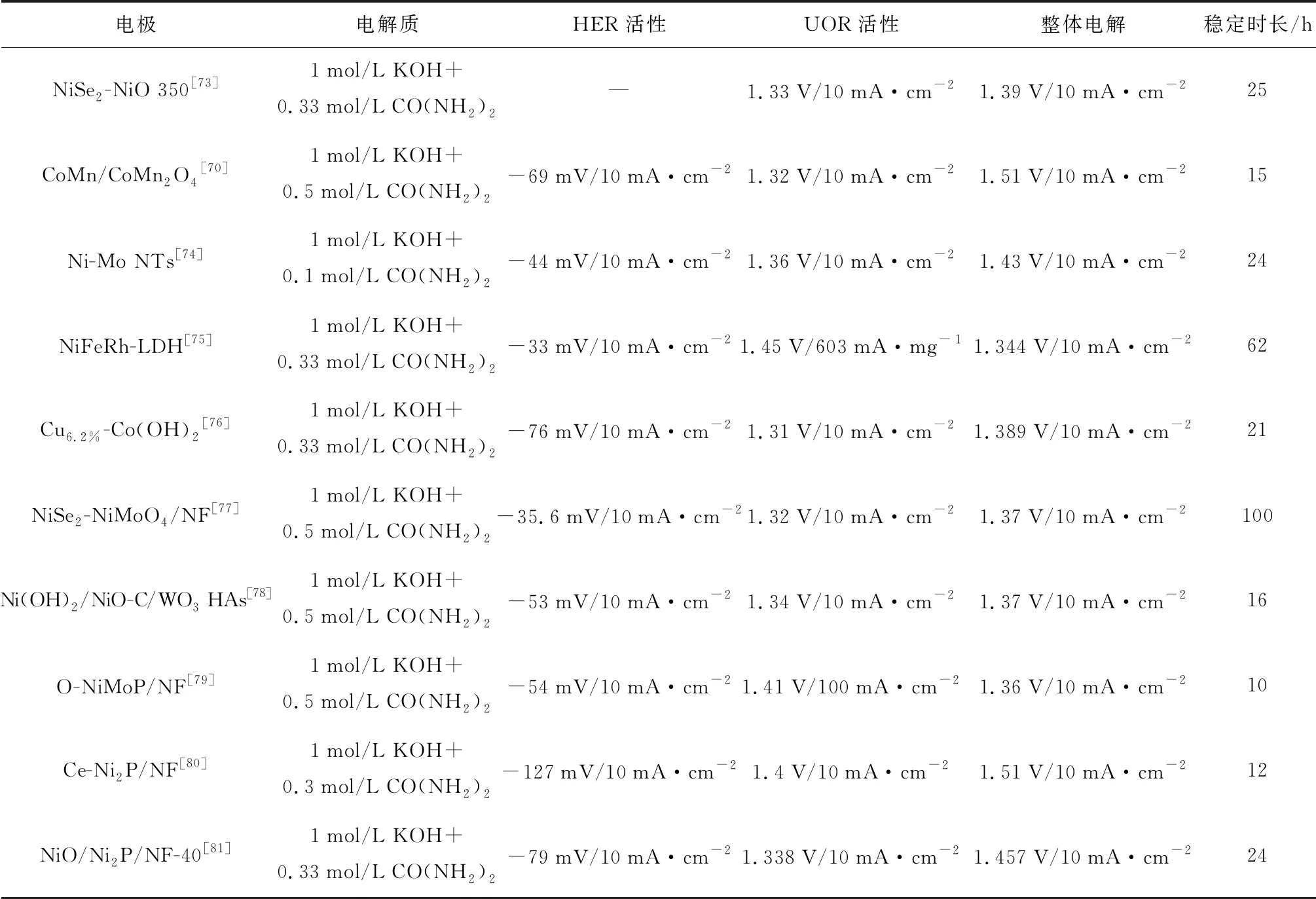
表4 用于尿素氧化反应与HER耦合的电催化剂及其活性
一般来说,碱性溶液中的尿素电解反应[69]如下。阴极反应
2H2O+2e-→ H2↑+2OH-
(6)
阳极反应
CO(NH2)2+6OH-→ N2↑+5H2O+CO2+6e-
(7)
总反应
CO(NH2)2+H2O → N2+3H2↑+CO2
(8)
UOR是一个六电子转移过程,动力学缓慢,导致其过电位增大,实际电池电压较高。因此,迫切需要一种具有高导电性和高活性的理想电催化剂,以增强其电催化活性[70-72]。早期的生物催化研究中,脲酶能够有效地加速尿素氧化分解为N2和CO2,并发现脲酶中含有两个Ni2+和相连的氢氧化物基团组成的活性位点[82-83]。Botte等[84-85]也发现,Ni在碱性介质中表现出比一些贵金属更好的UOR性能,并提出作用于UOR的Ni基催化剂的活性物种是NiOOH,而Ni基催化剂上的UOR遵循电化学氧化-化学氧化(E-C)机制。电化学氧化
Ni(OH)2+OH-→ NiOOH+H2O+e-
(9)
化学氧化
CO(NH2)2+6NiOOH+H2O → N2↑+
CO2↑+6Ni(OH)2
(10)
Xu等[86]开发了一种分层3D电催化剂(Ni12P5/Ni-Pi/NF),该催化剂由排列在NF上的Ni12P5和超薄非晶磷酸镍(Ni-Pi)纳米棒阵列组成。以Ni12P5/Ni-Pi/NF作为双功能催化剂构建的电解槽可以在1 mol/L KOH和0.5 mol/L CO(NH2)2中,以1.662 V的电压提供500 mA·cm-2的电流密度,与OER过程(1.949 V/500 mA·cm-2)相比,电压降低了约15%。Sun等[72]采用乙二醇辅助水热法制备了生长在泡沫镍上的Rh掺杂的NiFe-LDH纳米片阵列(NiFeRh-LDH/NF)。Ni和Fe的协同效应以及Rh掺杂导致的电子结构变化使得NiFeRh-LDH/NF具有出色的电催化反应动力学,在UOR与HER耦合的电解槽中,用1.344 V的电池电压即可实现10 mA·cm-2的电流密度,并在工作近100 h后电压仍保持稳定。Yu等[74]合成了圆球花状的NiSe2-NiMoO4异质结构催化剂,应用于整体水电解需要1.62 V的电池电压才能达到50 mA·cm-2的电流密度,加入0.5 mol/L CO(NH2)2后达到相同的电流密度所需电压仅为1.42 V。
元素掺杂、构建异质结构等方法除了可以调节电子结构、减小反应界面阻力之外,还可以借此引入丰富的空位,增加活性位点,优化中间物种的吸附能。参与UOR的含氮中间体中的吸附原子是N原子,开发氮空位稳定的金属氮化物能够有效地提高UOR活性。Li等[87]通过水热法在碳布上原位生长Ce-Ni(OH)2@CC,然后在NH3/Ar气氛下退火生成Ce-Ni3N@CC。合成的纳米片具有丰富的孔隙结构,将Ce掺杂到Ni3N中,可以为Ni3N提供丰富的氮空位和较大的比表面积,降低其电荷转移电阻并调节CO2吸附能,由此实现优异的UOR性能。与1 mol/L KOH中的OER活性相比,同样驱动10 mA·cm-2的电流密度,该电极的阳极电位在加入0.5mol/L CO(NH2)2后急剧下降,表明Ce-Ni3N@CC催化UOR反应的有效性,这种有效性在其他电极面前也不遑多让。以Ce-Ni(OH)2@CC为双功能电极构建混合电解槽,在10 mA·cm-2的电流密度下电池电压低至1.34 V,并在15 h之后仍能保持良好的电流密度。该实验展示了Ce-Ni3N@CC的高性能,表明UOR辅助制氢具有良好的发展前景。
尽管镍基复合材料在UOR反应中具有很大的优势,但其仍存在着耐久性差、易中毒、电导率低等问题,因此也有其他过渡金属基催化剂(Mn,Cu,Fe等)应用于UOR反应[70, 73]。而且,目前对于尿素电氧化的机理仍然不甚明确,缺乏更详细的解释,距UOR取代OER辅助制氢的实际应用还有很长一段路要走,探究更稳定、选择性更高的催化剂将是发展UOR与HER耦合的重要方向。
3.3 其他含氮化合物氧化反应与HER耦合
除肼和尿素以外,其他一些含氮化合物也可以用于与HER耦合以降低制氢能耗,如胺、液氨等化合物。
胺的热力学平衡电位为-0.77 V,远小于纯水电解的理论电位,极具热力学优势。胺通过电化学氧化生成亚胺、腈类和偶氮化合物等高价值化学品,如苯甲腈是药物、杀虫剂等精细化学品的常见前体[88]。胺的普通化学氧化需要苛刻的反应条件、有毒有害试剂[89]。与之相比,胺的电化学氧化更经济环保。Zeng等[90]设计了具有Se空位和Ni取代的原子厚度的CoSe2亚纳米带(CoSe2/Ni-SVs SBs)作为阳极催化剂,以CoP作为阴极催化剂,通过电催化氧化丁胺可以在1.37 V的电压下实现20 mA·cm-2的电流密度,氢气和腈类的法拉第效率分别达98.9%和96.7%。
液氨是一种非常好的能源载体,具有17.7%(质量分数)的储氢容量和3 000 W·h·kg-1的能量密度[91],氨的电化学氧化可以用于农业废水无害化处理,在反应中仅生成氮气和水,与阴极析出的氢气无混合爆炸的风险,且不会对环境造成影响。Zhang等[92]研究了一种锶和铜双掺杂的La2NiO4催化剂(La0.5Sr1.5Ni0.9Cu0.1O4-δ-Ar, LSNC-Ar)用于氨的电解和析氢反应耦合。该催化剂在HER过程中,驱动20 mA·cm-2的电流密度需要-1.45 V(以 Ag/AgCl电极作为参比电极)的电压。使用废水和0.5 mol/L KOH作为电解质,在1.22 V的低电压下工作170 h后,氨的去除率可以达到95%,展示了氨的电解与析氢耦合的前景。
然而,在已有的研究中,胺或氨的电化学氧化仍以贵金属基催化剂为主,在实际应用中这些反应仍存在很大的局限性,如电流密度不够稳定、催化剂不够经济实用等,还需要在未来的研究中逐一解决。
4 硫化物氧化反应与HER耦合
硫化物是一种具有毒性和腐蚀性的污染物,广泛存在于自然界(如H2S)和工业废水中[93],电化学氧化硫化物无需投入氧化剂,是治理硫化物污染十分高效的一种策略。硫离子作为电子供体提供电子的氧化反应(SOR)[94]如下
S2-→ S+2e-
(11)
SOR理论电位为-0.48 V,比OER更有利于热力学,与HER耦合更加经济可行。Gao等[95]设计了Ni、Co基硫化物包覆的Ni掺杂碳酸钴氢氧化钾针状纳米棒阵列(Ni-Co-S/NF)作为SOR催化剂,同时构建了一种不对称的酸碱耦合电解系统。阴极在酸性条件下析氢,阳极在碱性条件下产硫,只需-0.274 V的低压即可达到100 mA·cm-2的电流密度,产氢法拉第效率达97%~98%,产硫法拉第效率为95%~98%。
SOR辅助制氢从理论上阴阳极均可得到产品,是取代OER的极佳反应,但与其他氧化反应相同,后续阳极产物的纯化提取及催化剂的进一步开发仍处于探究中。
5 结论与展望
本文总结了目前可用的污染物(醇类、酚类、醛类、尿素、肼等)氧化降解与析氢反应耦合的研究进展。目前,关于尿素和肼的相关研究居多,前者催化剂研究丰富,后者理论氧化电位低,均可被完全矿化,无增值产品选择性反应、提纯等问题,用于工业化的可能性较高。其他污染物降解耦合制氢相对整体水电解也具有极大发展潜力。对这些耦合反应的研究除开发新型催化剂之外,还可以从构建混合电解系统的角度出发,提高电子利用率、防止O2/H2混合,实现低能耗制氢。此方法不仅可以绿色产氢,还能够在制氢的同时降解污染物,甚至生成更有价值的化学品。
但是,电解水制氢与治污的耦合还在发展当中。为实现应用,还有许多问题需要进行深入研究,具体如下。
(1) 设计开发成本低性能好的电催化剂。电极的研究可以通过改变衬底材料、控制形貌、调节构成、运用元素掺杂以调整中间体的电子结构和结合能、缺陷工程等方法来增强导电性,增加活性位点。尽管已经研究出多种过渡金属基催化剂,但贵金属基催化剂仍然是目前研究和应用的重点,这使得阳极氧化反应辅助制氢的设计因贵金属成本过高、储量较少等原因难以实现商业化。目前已有的研究表明,过渡金属基催化剂具有广阔的发展前景,性能优良、廉价易得,但相对于贵金属,性能方面仍然有所欠缺,稳定性也有待提高。此外,过渡金属在工业应用上能否适应大电流密度、阳极反应过程中的产物是否会引起电极中毒、电解液的pH与电极能否适配并保持稳定等,也是电极发展中亟需解决的问题。
(2) 优化混合电解槽。电解槽的设计与优化主要集中在其安全性、经济性和高效运行方面。首先,传统水电解槽为隔绝两边气体需要在阴阳极室之间设置隔膜,隔膜的腐蚀与更换不利于电解槽反应,且隔膜成本较高,不够经济环保。但是,在混合电解槽中,为适应阳极氧化反应构建特殊电解槽,膜的作用可以大幅度降低。其次,混合电解槽的设计应考虑阴阳极电解不同的物质而造成的阴阳极室电解液浓度不均的影响,注意阳极反应物的添加或反应过程的搅拌,以将影响降至最低。最后,若阳极产物是气体(如电解尿素生成N2),应根据产物特性设置相应的收集装置和氢气纯化装置,为投入实际生产提供可行性。
(3) 调节电解液pH。目前,酸碱性条件下的析氢都得到了很好的发展。由于电极在不同介质中具有不同的活性差异,因此针对不同的pH应有不同的设计。然而,大多数污染物的电化学氧化反应均发生在碱性环境中,而HER的半反应在酸性条件下活性更强。而且随着反应的进行,电解液pH也会发生变化,这使得电解液pH的调节十分有必要,对此可以建立阴阳极室酸碱不同的电解槽来进行反应。
(4) 探索更有利的阳极氧化反应。取代OER过程的阳极氧化反应一定相比电解水制氢能耗更低,可以降低反应成本,提高经济价值。氧化中间产物应不影响电极的活性,且能够实现大规模应用。其中,阳极氧化介质的溶解度、稳定性和电子转移动力学是决定其实际应用潜力的关键因素。除本文中提到的醇类、尿素等可以用作阳极氧化物之外,木糖[96]、葡萄糖[97]等生物质在混合水电解中也有应用。有些阳极反应甚至能够同时生产高价值的化学品,这些阳极反应产物的高选择性也应该得到关注,并考虑随后的分离和纯化。
(5)反应的催化机理。阳极氧化反应的生成物多种多样,探究反应的催化机理可以控制反应过程,提高对产物的选择性。应从催化剂的形貌、组成、活性位点及其在阴阳极的活性和稳定性等方面进行机理探究。在反应中,活性位点的确定对构建催化剂、解释催化过程具有重要意义,然而真实活性位点的确定非常困难,可使用原位方法,如原位拉曼光谱和原位傅里叶变换红外光谱(FTIR)联合SEM、TEM、XPS等测试,以识别真正的活性位点并监测反应的中间体。同时,可以通过DFT计算作为辅助从理论上推测反应机理。
(6)技术经济分析。不同于纯水电解制氢,混合水电解制氢以废水作为电解质,可以以各种工厂的工业废水、施肥所致的农业废水等作原料,从用料方面降低制氢的成本。醇类、醛类等化合物还可以在阳极生成酸盐或脂,在过程中增加除氢气以外的价值。污染物降解以达到排放标准,从结果上减少污水处理的花费,即是一种增收。但是,用于阳极氧化的污染物分子量一般大于O2,所产氢气每个质子需要一个电子,产生同样氢气的量所需有机物要更多[98],在工业化方面不占据优势。一些污染物如尿素完全矿化会造成碳排放,产生环境成本。在阳极生成增值产品后,后续的分离、纯化也需要额外投入。加之电极开发与定期更换、废水运输集中等成本,从经济可行性角度分析,电化学阴极制氢耦合阳极污染物降解距商业化应用仍需很多努力。
