基于4个自噬相关LncRNA表达信息学分析构建肺鳞癌预后风险评分模型
2023-12-14吴瑶何杰张维
吴瑶,何杰,张维
(成都医学院第一附属医院呼吸与危重症医学科,四川 成都 610500)
肺鳞状细胞癌(肺鳞癌)属于肺癌常见的病理亚型之一[1-2]。虽然近年来对于肺鳞癌病人的治疗取得了较大进步,但病人的5年生存率仍然没有超过20%[3-4]。构建一种新的并且有效的预测模型将有助于肺鳞癌病人的预后判断和个体化治疗。自噬是真核生物进化中一种高度保守的生物学过程[5],自噬和自噬相关基因的调节在肿瘤的发生发展中发挥着重要作用[6]。YANG等[7]对人体肝癌组织和正常肝脏组织对比研究发现,HOX转录物反义RNA(HOTAIR)在肝癌组织中表达量显著高于正常肝组织,并且同时伴随着自噬基因的高表达,提示在肿瘤细胞中,自噬的强化可能与基因自噬相关长链非编码RNA(LncRNA)过度表达密切相关。高德军等[8]研究表明,LncRNA可通过靶向调节miR-140-5p促进肺癌细胞的增殖和自噬,抑制凋亡。鉴于LncRNA在肺癌自噬中的重要作用,推测自噬相关LncRNA可能可以作为肺鳞癌的预后标志物。本文旨在用生物信息学方法研究自噬相关LncRNA对肺鳞癌的预后价值并构建预后模型,为肺鳞癌病人的预后评估提供一定参考。
1 资料和方法
1.1 研究对象
从癌症基因组图谱(TCGA)官方网站(网址为https://gdc-portal.nci.nih.gov/)下载肺鳞癌病人肿瘤组织和癌旁组织的mRNA测序数据及临床信息。通过Illumina高通量测序平台获得mRNA测序数据。下载的数据内容包含肺鳞癌病人502例肿瘤组织的mRNA表达数据和49例癌旁正常组织的mRNA表达数据。临床数据包含病人的ID号、年龄、性别、病理分期、生存时间、生存状态等,为了减少统计学误差,剔除生存时间少于30 d的病人信息,最终纳入482例肺鳞癌病人。
1.2 自噬相关LncRNA的提取
从HADb(http://autophagy.lu/)数据库中获得自噬基因232个,通过ActivePerl软件(版本号5.26)提取232个自噬基因在TCGA的表达谱并分类出LncRNA表达谱。使用R软件中的“Limma”包(https://www.R-project.org)对mRNA测序表达值进行log2转换,以|log2(fold change)|>1.5和伪发现率(FDR)<0.05为阈值,对数据进行归一化和差异表达基因分析。对筛选出的差异表达的自噬基因进行聚类分析,并绘制热图。使用R软件中的“corrplot”包筛选出与自噬基因相关的LncRNA,阈值设定为相关系数(r)绝对值>0.5和P<0.05。
1.3 实验验证
1.3.1标本来源 随机选取成都医学院第一附属医院2016年3月-2019年7月收治的50例肺鳞癌病人手术切除的癌组织及其对应的癌旁正常组织样本,术后病理结果均证实为肺鳞癌。所有的组织样本获取均由病人或家属签署知情同意书,并获得成都医学院第一附属医院伦理委员会批准。
1.3.2引物设计和合成 所有引物的设计和合成均由杭州齐步生物工程(浙江)股份有限公司完成,以GAPDH为内参基因,引物及其序列见表1。

表1 4个LncRNA和GAPDH基因的引物序列
1.3.3实时定量PCR检测RNA 按照PCR试剂盒使用说明书方法提取组织内总RNA并逆转录为cDNA,应用SYBR®Premix Ex TaqTM10 μL进行RT-qPCR,反应在ABI 8000实时定量PCR仪进行,以GAPDH为内参,上游引物(10 μmol/L)、下游引物(10 μmol/L)均0.4 μL,cDNA 1 μL,RNase Free Water 8.4 μL。反应条件设定为:95 ℃预变性30 s;95 ℃、5 s,60 ℃、34 s,共40个循环;60 ℃退火30 s。以2-△△ct表示RNA的相对表达量。
1.4 统计学分析

2 结 果
2.1 纳入病人的基本情况
符合纳入标准的肺鳞癌病人482例,其中男347例,死亡235例;女135例,死亡94例。肺鳞癌死亡病人平均年龄(68.7±6.9)岁,未死亡病人平均年龄(65.4±8.5)岁,两组年龄比较差异无显著性(t=1.79,P>0.05)。病理分期Ⅰ、Ⅱ期与Ⅲ、Ⅳ期病人数比较(χ2=3.64,P<0.05)、有无化疗病人例数比较(χ2=6.62,P<0.05)、生存时间比较(t=2.98,P<0.05)差异均有显著性。见表2。

表2 从TCGA数据库中下载纳入分析的肺鳞癌病人的基本特征(n=482)
2.2 肺鳞癌组织和正常肺组织中差异表达的自噬基因
对502例肺鳞癌病人肿瘤组织和49例癌旁正常肺组织的自噬基因差异分析显示,满足条件的自噬基因共有30个。对这些基因进行聚类分析,并绘制热图(图1),结果显示高表达基因有20个,低表达基因10个。
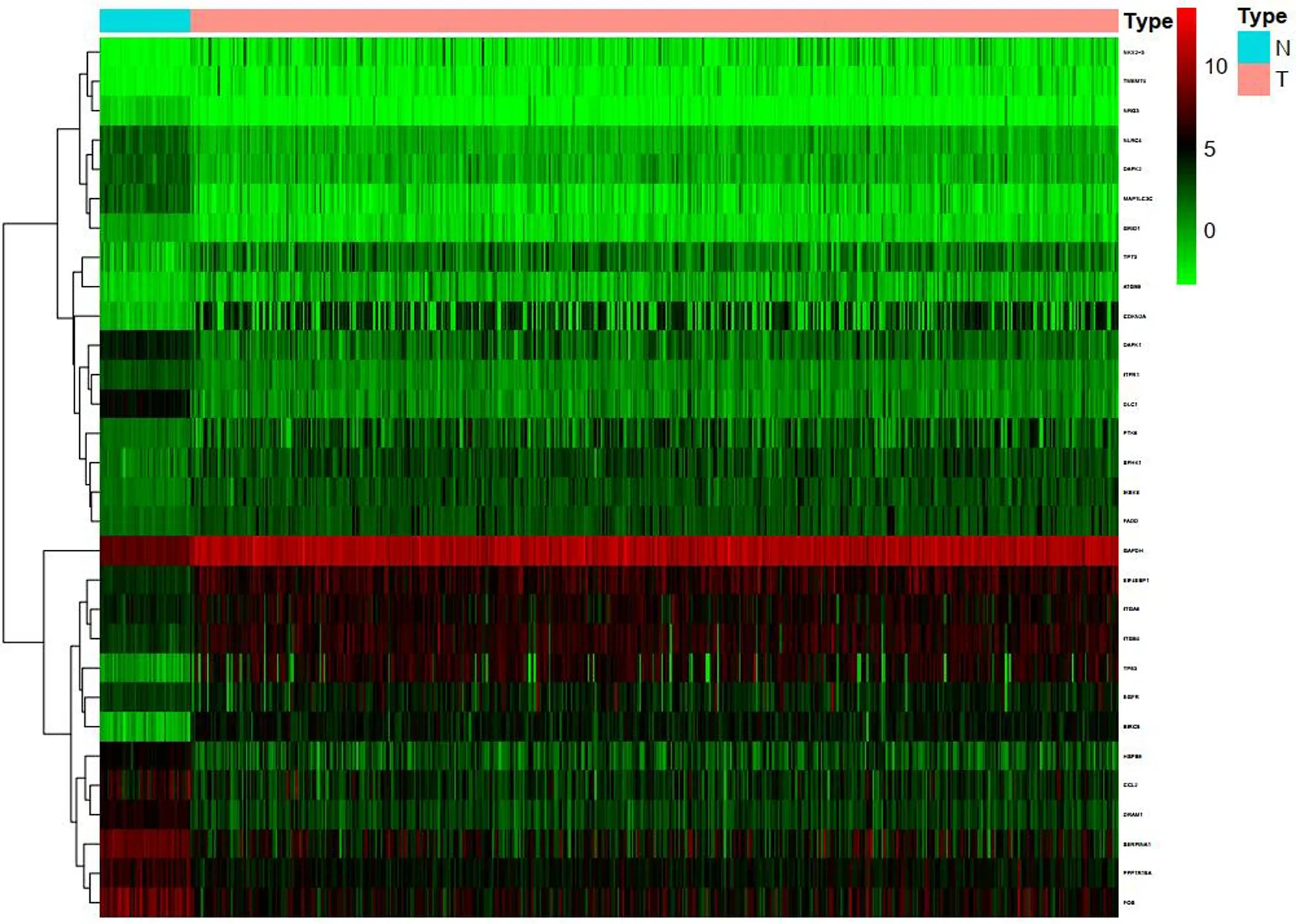
N:正常组织;T:肺鳞癌组织。
2.3 自噬相关LncRNA的确定
通过Perl软件共提取出14 142个LncRNA,根据筛选条件,获得自噬相关LncRNA有89个,结合TCGA来源对应的临床信息,通过单因素Cox回归分析,初步筛选出与肺鳞癌预后相关的LncRNA有16个;通过LASSO回归分析进一步确定了4个关键的自噬相关LncRNA,它们分别是AL365356.4、AC012181.1、AL390719.2、AC245060.2。4个自噬相关LncRNA的详细信息见表3。
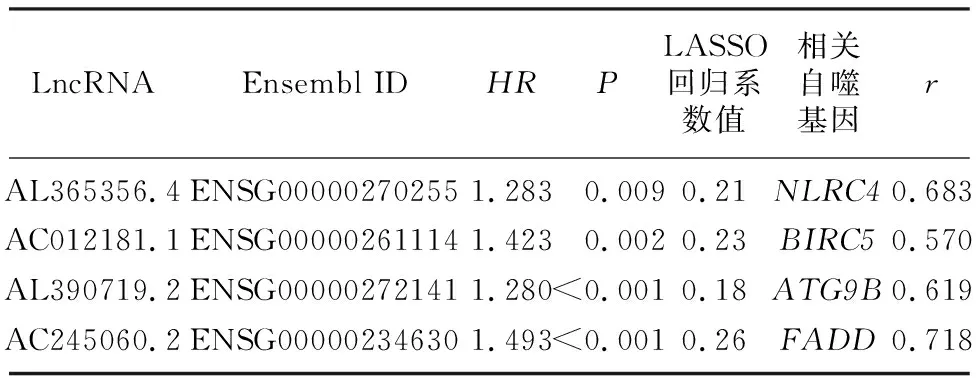
表3 4个自噬相关LncRNA的详细信息
2.4 肺鳞癌预后风险评分模型构建和评价
基于LASSO回归分析确定的4个自噬相关LncRNA构建预后风险评分模型,其风险评分=0.26×AC245060.2+0.18×AL390719.2+0.23×AC012181.1+0.21×AL365356.4。K-M生存分析显示,低风险组与高风险组生存曲线存在差异,低风险组生存率较高(χ2=15.267,P<0.05)(图2A)。ROC曲线分析显示,3、5年AUC分别为0.746和0.743(图2B)。同时,高风险组病人4个LncRNA呈现更高的表达水平(图3A);风险评分分布及风险评分与生存时间的关系分析提示高风险组病人总生存时间更短(图3B、C)。以总生存时间为因变量,预后模型所计算的风险评分、年龄、性别、病理分期作为协变量进行多因素Cox回归分析,结果表明风险评分可以作为独立的预后预测因子(HR=2.100,95%CI=1.541~2.861,P<0.01)。见图4。
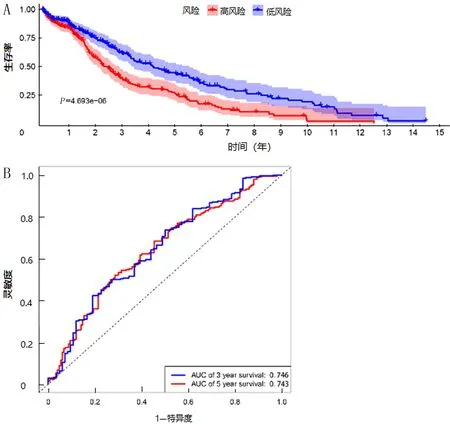
A:高风险和低风险病人的生存分析图;B:预后模型的3年和5年ROC曲线。
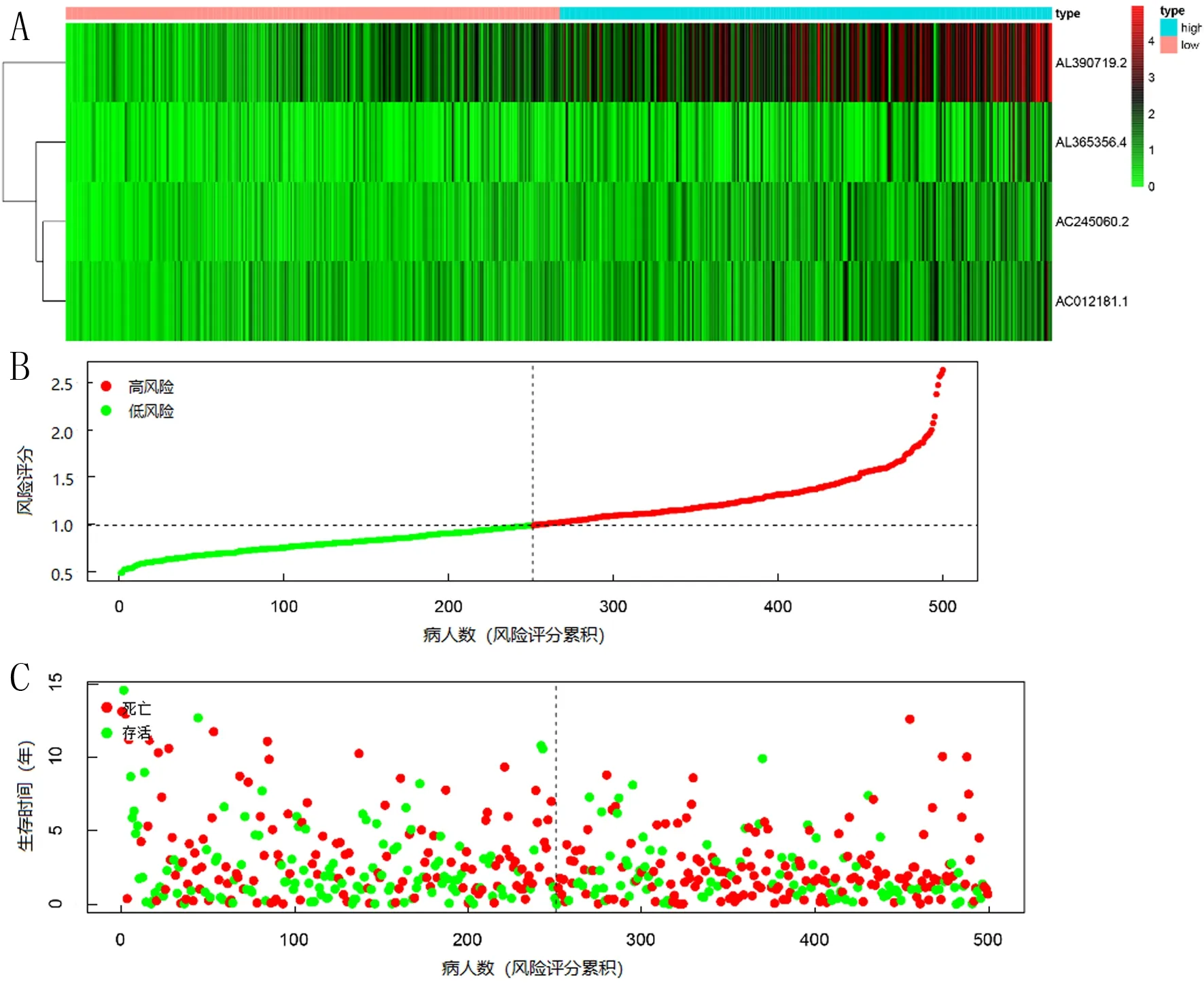
A:高风险和低风险肺鳞癌病人AL365356.4、AC012181.1、AL390719.2、AC245060.2的表达谱热图;B:高风险病人和低风险病人的风险评分分布;C:高风险病人和低风险病人的生存状态分布。

图4 多因素Cox回归分析森林图
2.5 基础实验验证结果
纳入肺鳞癌病人50例,男35例,女15例;年龄28~76岁,中位年龄47岁;TNM分期Ⅰ期20例,Ⅱ期15例,Ⅲ期10例,Ⅳ期5例。RT-qPCR检测显示,肺鳞癌组织AC245060.2表达水平为3.391±0.275,正常组织0.692±0.016,两组比较差异有显著性(t=69.28,P<0.05);肺鳞癌组织AL390719.2表达水平为1.842±0.054,正常组织0.061±0.021,两组比较差异有显著性(t=17.35,P<0.05);肺鳞癌组织AC012181.1表达水平为0.867±0.179,正常组织0.117±0.061,两组比较差异有显著意义(t=28.04,P<0.05);肺鳞癌组织AL365356.4表达水平为4.786±0.681,正常组织1.064±0.231,两组比较差异有显著性(t=36.59,P<0.05)。
3 讨 论
LncRNA为一种转录本长度大于200个核苷酸的非编码RNA,缺乏完整的特异性开放阅读框,且无编码蛋白质的功能[9-10]。大量研究结果表明,LncRNA可以通过组蛋白修饰、染色质重塑、RNA代谢等诸多生物学过程,在表观遗传、转录以及转录后水平调控自噬基因的表达[11]。LncRNA可以发挥内源性miRNA海绵功能与miRNA相互作用,参与对自噬基因的表达调控;同时,miRNA也可以通过RNA诱导沉默复合物调控LncRNA发挥生物学功能,两者又可竞争结合mRNA,共同参与肿瘤的发生和发展过程[12-13]。
目前,许多证据表明LncRNA的过表达、缺失或是突变可以通过调控肿瘤的自噬基因而对肿瘤的恶性生物行为产生驱动作用。对长链非编码肺癌转移相关转录本1(LncRNA MALAT-1)的研究显示,LncRNA MALAT-1下调可以抑制自噬活性,从而抑制A549肺癌细胞的增殖和扩增,促进肺癌细胞的凋亡。此外,一些异常的LncRNA也与肺癌的不良预后密切相关,有研究表明,LncRNA肌动蛋白纤维相关蛋白1-反义RNA1(AFAP1-AS1)在非小细胞肺癌组织中的表达量升高,且高表达AFAP1-AS1的肺癌病人更容易出现淋巴结转移,总生存时间更短[14]。尽管目前关于LncRNA在肺癌自噬中的研究已有很多,但多数是单个LncRNA对自噬的调节作用,范围较局限,自噬相关LncRNA在肺鳞癌中的作用和机制以及在临床预后评估中的应用仍然有待进一步探索。
本研究通过检索在线数据库,下载肺鳞癌自噬相关LncRNA表达数据和病人的临床特征,通过生物信息分析发现,在肺鳞癌中有许多自噬基因相关的LncRNA的表达发生了异常改变,这些基因可能与肺鳞癌的发生、进展以及预后相关。本文初步筛选出了自噬基因相关的LncRNA有89个;进一步进行单因素Cox回归分析,共筛选出16个自噬相关LncRNA和临床预后密切相关。但单因素Cox回归分析每次仅纳入一个变量,可能存在过度拟合的现象,因此本文再次引入LASSO回归分析进行降维。LASSO回归分析的特点是在拟合广义线性模型的同时进行变量筛选和复杂度的调整,有选择性地把变量放入模型以得到更好性能参数,并通过一系列的参数控制模型的复杂度,从而避免产生过度拟合,这样很好地弥补了单因素Cox回归分析的不足。本文通过LASSO回归分析,最终确定了4个关键的自噬相关LncRNA构建模型,该模型可区分高风险组和低风险组的病人,而且预测性能较好。最后,多因素Cox回归分析显示,风险评分和预后相关,且具有预测独立性。为了更进一步验证模型中的4个LncRNA的临床意义,本研究收集了50例肺鳞癌病人的肿瘤组织标本,检测其LncRNA的表达量,结果显示上述4个LncRNA在肺鳞癌组织中的表达水平均高于正常组织,提示这4个自噬相关LncRNA高表达病人可能预后更差。
遗憾的是,虽然本研究结果显示模型中的4个LncRNA与肺鳞癌预后密切相关,且风险评分可以作为独立的预后因子,但是目前尚缺乏其临床研究或者基础实验研究,它们在肺鳞癌发生中的作用机制仍不清楚,有待进一步探索。另外,本研究采用的生物信息分析的方法和工具较多,利用系统的方法处理大量的数据是其优势,但仍然存在一定的不足之处:①大部分数据均来自于TCGA数据库,未通过其他数据库如GEO验证;②预后风险评分模型仅纳入了自噬相关LncRNA表达水平,未考虑其他基因改变,如小RNA、环状RNA等表达水平改变对预后的影响。在下一步研究中,我们将结合自己的验证数据和随访信息开展更为深入的生物学水平的机制研究;同时,考虑纳入更多可能影响临床预后的因素,如吸烟情况、其他非编码RNA表达水平等,以期构建更为稳定和可靠的预后模型。
综上所述,本研究通过分析TCGA数据库信息,构建了一个基于4个自噬相关LncRNA的肺鳞癌预后模型,该模型为肺鳞癌预后的评估提供了有意义的参考。
