Histone deacetylase inhibitor pracinostat suppresses colorectal cancer by inducing CDK5-Drp1 signaling-mediated peripheral mitofission
2023-12-14XioLingLingLnOuyngNnNnYuZhengHuSunZiKngGuiYuLongNiuQingYuHeJingZhngYngWng
Xio-Ling Ling ,Ln Ouyng ,Nn-Nn Yu ,Zheng-Hu Sun ,Zi-Kng Gui ,Yu-Long Niu ,Qing-Yu He ,*,Jing Zhng ,Yng Wng ,*
a MOE Key Laboratory of Tumor Molecular Biology and Key Laboratory of Functional Protein Research of Guangdong Higher Education Institutes,Institute of Life and Health Engineering, College of Life Science and Technology, Jinan University, Guangzhou, 510632, China
b Department of Radiology, The First Affiliated Hospital of Jinan University and MOE Key Laboratory of Tumor Molecular Biology, Jinan University,Guangzhou, 510632, China
Keywords:HDAC inhibitor Pracinostat CDK5 Mitochondrial fission Acetylation Drp1
ABSTRACT Divisions at the periphery and midzone of mitochondria are two fission signatures that determine the fate of mitochondria and cells.Pharmacological induction of excessively asymmetric mitofissionassociated cell death (MFAD) by switching the scission position from the mitochondrial midzone to the periphery represents a promising strategy for anticancer therapy.By screening a series of paninhibitors,we identified pracinostat,a pan-histone deacetylase (HDAC) inhibitor,as a novel MFAD inducer,that exhibited a significant anticancer effect on colorectal cancer (CRC) in vivo and in vitro.Pracinostat increased the expression of cyclin-dependent kinase 5(CDK5) and induced its acetylation at residue lysine 33,accelerating the formation of complex CDK5/CDK5 regulatory subunit 1 and dynaminrelated protein 1 (Drp1)-mediated mitochondrial peripheral fission.CRC cells with high level of CDK5(CDK5-high) displayed midzone mitochondrial division that was associated with oncogenic phenotype,but treatment with pracinostat led to a lethal increase in the already-elevated level of CDK5 in the CRC cells.Mechanistically,pracinostat switched the scission position from the mitochondrial midzone to the periphery by improving the binding of Drp1 from mitochondrial fission factor (MFF) to mitochondrial fission 1 protein(FIS1).Thus,our results revealed the anticancer mechanism of HDACi pracinostat in CRC via activating CDK5-Drp1 signaling to cause selective MFAD of those CDK5-high tumor cells,which implicates a new paradigm to develop potential therapeutic strategies for CRC treatment.
1.Introduction
Mitochondria are well-known as key platforms for cell signaling cascades and play a fundamental role in energy production.To adapt to changing stressors,mitochondria are highly dynamic,switching between fusion and fission status in order to maintain a functional mitochondrial network [1].In mitochondrial fission,dynamin-related protein 1 (Drp1) is activated by phosphorylation at Ser-616 and translocated from the cytosol to the mitochondria by binding to different adaptor proteins on the mitochondrial surface,such as mitochondrial fission factor(MFF)or mitochondrial fission 1 protein (FIS1) [2].Two types of mitochondrial fission (mitofission),periphery and midzone divisions,result in distinct mitochondrial fates.Scission at the midzone-mediated by Drp1-MFF interaction results in mitochondrial proliferation and tumorigenesis [3],whereas peripheral fission regulated by Drp1-FIS1 interaction is linked to mitochondrial dysfunction,mitophagy,and cell death[4].Multiple studies have found that excessively asymmetric mitochondrial division initiates cell death and is thus lethal for cancer cells,suggesting that inducing asymmetric mitofissionassociated cell death (MFAD) may be a promising strategy for cancer therapy [5].
Histone deacetylase inhibitors (HDACis) are emerging as new anticancer drugs that regulate DNA damage,autophagy,and cell death via epigenetic or non-epigenetic mechanisms [6,7].Vorinostat,romidepsin,belinostat,and panobinostat are four HDACis that have been approved by the US Food and Drug Administration (US FDA)for the treatment of skin/peripheral T-cell lymphoma,multiple myeloma[8],and colorectal cancer(CRC)[7,9].Many HDACis induce cell apoptosis in a mitochondria-dependent manner;however,whether HDACis modulate mitochondrial dynamics remains unclear.Pracinostat is an orally active pan-HDACi with significant anticancer activity [10] that possesses excellent drug properties,including good absorption,distribution,metabolism,excretion,high bioavailability,and safety[11].Pracinostat suppresses the metastasis and proliferation of breast cancer by inhibiting the interleukin 6/signal transducer and activator of the transcription 3 (STAT3)signaling pathway [12].However,specific biomarkers in cancers that effectively respond to pracinostat remain elusive.Therefore,a systematic understanding of the action mechanism of pracinostat has the potential to be of great clinical value by providing a new paradigm for developing more effective cancer treatment strategies.
Cyclin-dependent kinase 5 (CDK5) is an atypical member of the cyclin-dependent kinase family that has been well studied for its role in cancer development.Its activity is dependent on non-cyclin activator CDK5 regulatory subunit 1 (CDK5R1/p35),which phosphorylates a variety of substrates,including STAT3.The kinase activity of CDK5 is regulated by post-translational modifications(PTM),such as phosphorylation,S-nitrosylation and acetylation[13].Accumulating evidence has demonstrated that CDK5 activation is critical for tumorigenesis[14];thus,relevant therapeutic strategies are urgently needed.Herein,we examined the effects of pracinostat on mitochondrial dynamics and tumor growth of CRC cells in vitro and in vivo.Confocal imaging,data independent acquisition(DIA)-based quantitative proteomics,and a series of functional assays were performed to illustrate that pracinostat exerts the anticancer effect through CDK5-Drp1 signaling-mediated mitochondrial peripheral fission.The findings provide insight into the anticancer activity of HDACi pracinostat in CRC with high CDK5 level,and represent a promising therapeutic strategy for CRC treatment.
2.Materials and methods
2.1.Cell culture and drugs
Human CRC cell lines HCT116 and HT29 (ATCC,Rockville,MD,USA) were cultured in Dulbecco's modified Eagle's medium(DMEM;Gibco,Waltham,MA,USA),containing 10% fetal bovine serum (Gibco) in a 37°C,5% CO2incubator.The pan-inhibitor library (20 compounds),roscovitine,SIS17,CI994,TMP195,vorinostat and trichostatin A were acquired from Topscience (Shanghai,China).
2.2.Plasmid and transfection
Full-length CDK5 and Drp1 were amplified and cloned into the lentiviral pLVX-MCS-Puro vector by using the Clone Express®II One Step Cloning Kit(Vazyme,Nanjing,China).The plasmid pLVX-CDK5-K33R was constructed using the Mut Express®II Fast Mutagenesis Kit(Vazyme) according to the manufacturer's instructions.Cells were transfected with pLVX-CDK5-flag,pLVX-CDK5-K33R-flag or vector control using Lipofectamine 3000 reagent(Invitrogen,Carlsbad,CA,USA)according to the manufacturer's instructions.
2.3.Cell viability assay
Cells were digested and seeded in 96-well plates,and then treated with drugs at different concentrations.Three replicate wells were used in each experiment,with approximately 3000 cells/well.Cell activity was measured by the CCK-8 assay (Topscience).The CCK-8 reagent was added into the cultured cells at 10% working concentration and incubated at 37°C for 1.5 h.Absorbance was measured at 450 nm using a microplate reader (BioTek Instruments,Winooski,VT,USA).
2.4.Colony formation assay
Cells were seeded in 12-well or 6-well plates,and three replicate wells were used in each experiment.Different drug concentrations were added.When the cells grew to form visible clone sites,the plates were gently washed with phosphate-buffered saline (PBS)twice,fixed with 4%paraformaldehyde(Leagene,Beijing,China)for 15 min and stained with 0.1%crystal violet for 10 min.After gently washing the crystal violet off the cell surface with water,the cells were photographed using scanner.
2.5.Flow cytometric analysis
Cells were seeded in 6-well plates and treated with different drug concentrations.Cell apoptosis was determined using an Annexin V FITC/PI Apoptosis Detection kit(KeyGen,Nanjing,China)and measured using a flow cytometer (BD Biosciences,San Diego,CA,USA).For mitochondrial membrane potential(MMP)detection,cells were stained with JC-1 (Beyotime,Shanghai,China) after pracinostat treatment and measured using a flow cytometer.The FlowJo software (BD Biosciences) was used to quantify the data obtained from the flow cytometer.
2.6.Western blot analysis
Cell lysates were prepared as previously described [15],and an equal volume of loading buffer was added and boiled.Protein samples were loaded onto a sodium dodecyl sulfatepolyacrylamide gel electrophoresis (SDS-PAGE) gel,electrophoresed,and subsequently electrotransferred to a polyvinylidene fluoride (PVDF) membrane (Millipore,Bedford,MA,USA).The membrane was incubated with the primary antibody overnight at 4°C,and then incubated with the corresponding secondary antibody for 1 h at room temperature.After washing with PBS three times,the PVDF membrane was visualized using the ECL substrate(Bio-Rad,Hercules,CA,USA).The primary antibodies used werep-Drp1 (Cell Signaling Technology,Danvers,MA,USA),panacetylation (PTM Biolab,Hangzhou,China),Drp1,CDK5R1,MFF,FIS1,MFN1,MFN2(Proteintech,Wuhan,China),caspase-3,cleaved caspase-3,cleaved-PARP,and CDK5(ABclonal,Wuhan,China),antiflag (Abbkine,Shanghai,China) and β-actin (Santa Cruz,CA,USA).
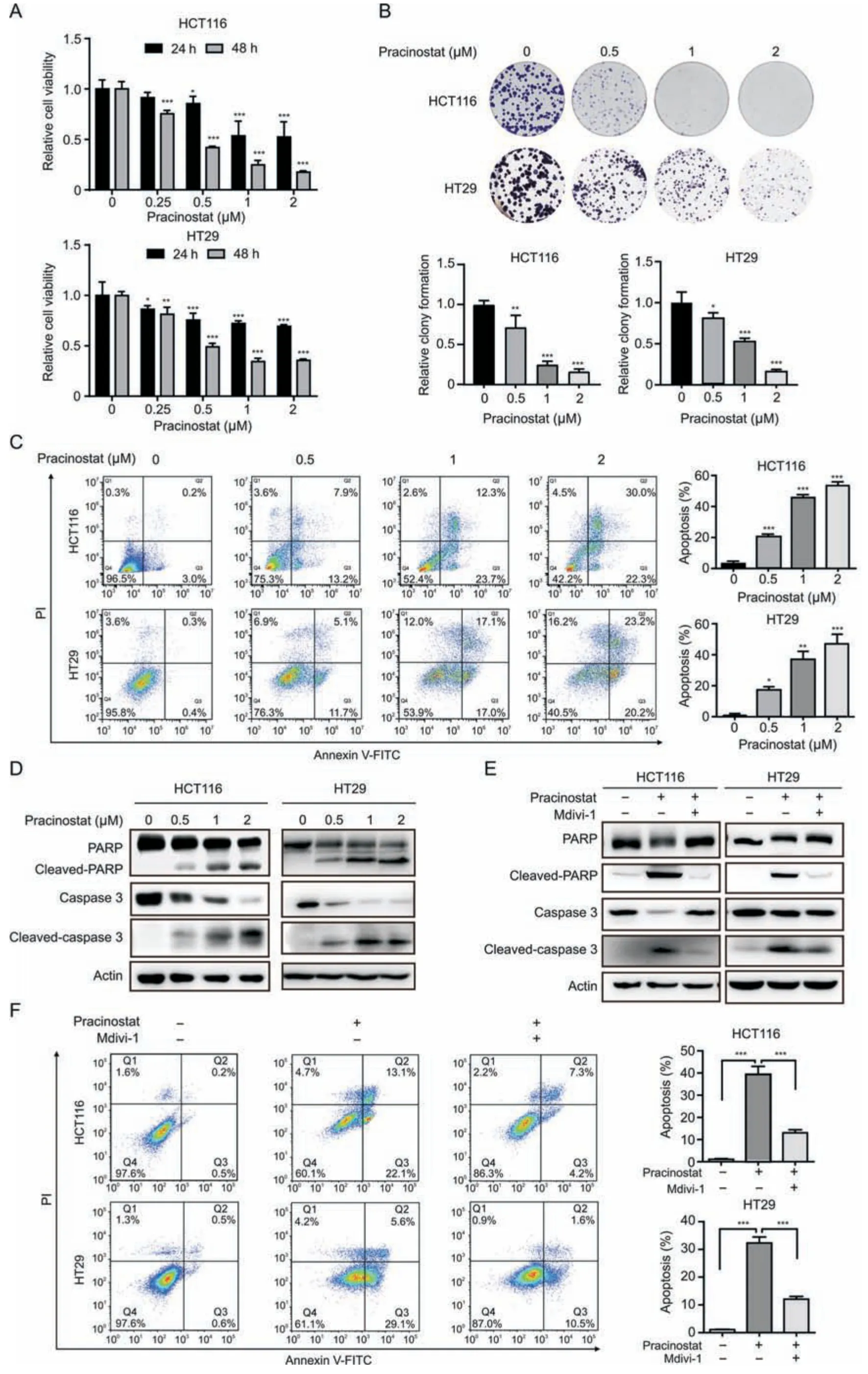
Fig.2.Pracinostat induces colorectal cancer(CRC)cell apoptosis in a dynamin-related protein 1(Drp1)-dependent manner.(A)The cell viability of HCT116 and HT29 cells treated with increased concentrations of pracinostat for 24 h and 48 h was determined by CCK-8 assay.(B) Comparison of the colony formation ability of both HCT116 and HT29 treated with pracinostat(0-2 μM)as indicated.(C)Apoptosis in HCT116 and HT29 cells treated with pracinostat(0-2 μM,48 h)was determined by the Annexin V/PI assay.The apoptotic cells with Annexin V positive were quantified.(D) Immunoblot analysis of the apoptosis markers (PARP,cleaved-PARP,caspase 3,and cleaved-caspase 3) in both HCT116 and HT29 cells treated with pracinostat as indicated.(E) Immunoblot analysis of the apoptosis markers (PARP,cleaved-PARP,caspase 3,and cleaved-caspase 3) in both HCT116 and HT29 cells treated with pracinostat and/or Mdivi-1 (5 μM) as indicated.(F) Apoptosis in HCT116 and HT29 cells treated with pracinostat (1 μM) and/or Mdivi-1 (5 μM) was determined by the Annexin V/PI assay.The apoptotic cells positive for Annexin V were quantified.Representative results were performed from three independent experiments;error bars,standard deviation (SD).P values were indicated by a two-tailed unpaired Student's t-test.*P <0.05,**P <0.01,***P <0.001 when compared with control.FITC:fluorescein isothiocyanate;PI: propidium iodide;PARP: poly (ADP-ribose) polymerase.
2.7.Mass spectrometry and bioinformatics analyses
Protein digestion and data independent acquisition (DIA)-based quantitative proteomics analysis were performed as previously described[5,16].In brief,HCT116 cells treated with pracinostat(1 μM)were lysed with lysis buffer (Cell Signaling Technology).Then,the proteins were digested with trypsin(Life,Gaithersburg,MD,USA)at 37°C for 16 h.Peptide fragments were vacuum freeze-dried,resuspended in anhydrous acetonitrile solution,and desalted with MonoTIP C18 (GL Sciences,Tokyo,Japan).The peptide samples were analyzed using an Orbitrap Fusion Lumos mass spectrometer(Thermo Fisher Scientific,Waltham,MA,USA).Raw data were analyzed using Spectronaut software (Omicsolution,Shanghai,China).Protein and peptide false discovery rate were set to 1%.Gene ontology(GO)and ingenuity pathway analyses(IPA;www.ingenuity.com,QIAGEN,Shanghai,China) were used to analyze differentially expressed proteins(DEPs).
2.8.Confocal assay
Approximately 1000 cells were seeded into a confocal Petri dish.After cell adhesion,different concentrations of drugs were added.After treatment,the dishes were stained with Mito-Tracker Red(Beyotime),Drp1 antibody (Proteintech),or 4’,6-diamidino-2-phenylindole (DAPI;Beyotime),and subsequently subjected to confocal laser microscopy (LSM900,Carl Zeiss,Jena,Germany) for imaging analysis.The mitochondrial reactive oxygen species(ROS)level was measured by staining MitoSOX Red Mitochondrial Superoxide Indicator (ABclonal) in HCT116 and HCT116-CDK5 cells.The cells before and after 30 min of pracinostat exposure were analyzed by confocal laser microscopy,and the fluorescence intensities of ROS in the mitochondria were compared.
2.9.Mitochondrial morphology analysis
Images of mitochondrial morphology obtained from confocal laser microscopy were analyzed using the MiNA plug-in of Image J software (National Institutes of Health,Bethesda,MD,USA) according to previous methods [17].Briefly,at least three random visual fields were selected for each condition,the relative number of tubular and fragmented mitochondria,and the mean rod/branch length per cell were evaluated.
2.10.Co-immunoprecipitation (Co-IP)
Immunoprecipitation was performed as previously described[18].Cell lysates were incubated with IgG (Santa Cruz Biotechnology) and protein A/G plus agarose beads (Santa Cruz Biotechnology) for 1 h.Then anti-flag (Abbkine) and IgG primary antibodies were mixed with the supernatant overnight at 4°C.Next,protein A/G plus agarose beads were added to the supernatant.The beads were centrifuged and washed with PBS,followed by mixing with 5 × SDS/PAGE loading buffer (Beyotime) for Western blot analysis.
2.11.Transmission electron microscopy (TEM)
Treated cells were harvested and fixed with an electron microscope fixative (Servicebio,Wuhan,China).The cells were washed with 0.1 M phosphate buffer (PB;pH 7.4) and fixed with 1% OsO4(Ted Pella,Inc.,Redding,CA,USA)in 0.1 M PB for 2 h.Subsequently,the cells were dehydrated using an alcohol gradient(30%,50%,70%,80%,95%,and 100%ethyl alcohol),and 100%acetone.Samples that underwent embedding,polymerization,ultrathin slicing,and staining were subjected to images capture.Observations for image acquisition were performed using TEM (HT7800,HITACHI,Tokyo,Japan)for the cuprum grids.The mitochondrial area was calculated using Image J software.
2.12.Tumorigenicity in nude mice
All animal experiments involving nude mice were approved by the Animal Experiment Ethics Committee of Jinan University and were conducted in accordance with institutional guidelines for animal welfare (Approval numbers: 20,200,717-01/20,211,116-12).Tumor cells were resuspended in PBS,mixed 1:1 (V/V) with matrigel (Corning,NY,USA),and subcutaneously injected into mice at a density of 1 × 106cells/mouse.Once the solid tumors reached a diameter of~5 mm,each mouse was administered 25 mg/kg of pracinostat or solvent every two days.The body weight and size of the transplanted tumors were measured every two days.The tumors were dissected and weighed before reaching a diameter of approximately 12 mm.Histological examinations (HE),immunohistochemical staining (IHC),Ki-67 staining,terminal deoxynucleotidyl transferase-mediated dUTPbiotin nick end labeling (TUNEL) staining assays,and serum biochemical tests were carried out for the assessment of therapy efficacy,according to the protocols described in our previous studies [19].
2.13.Immunohistochemical staining
Paraffin sections of transplanted tumor tissues were processed by dewaxing and hydration through a graded series of ethanol solutions.After treatment with 3%hydrogen peroxide in methanol solution,the resulting slides were placed in a citrate buffer(pH =6)and boiled for 10 min.The slides were then rinsed twice and blocked with goat serum for 1 h.Ananti-Ki67 primary antibody(Proteintech) was added,and the sections were incubated overnight,followed by exposure to the corresponding secondary antibody solution for 1 h.Finally,the slides were washed and sealed with neutral resin before being photographed under a microscope(DS-Ri2,Nikon,Tokyo,Japan).
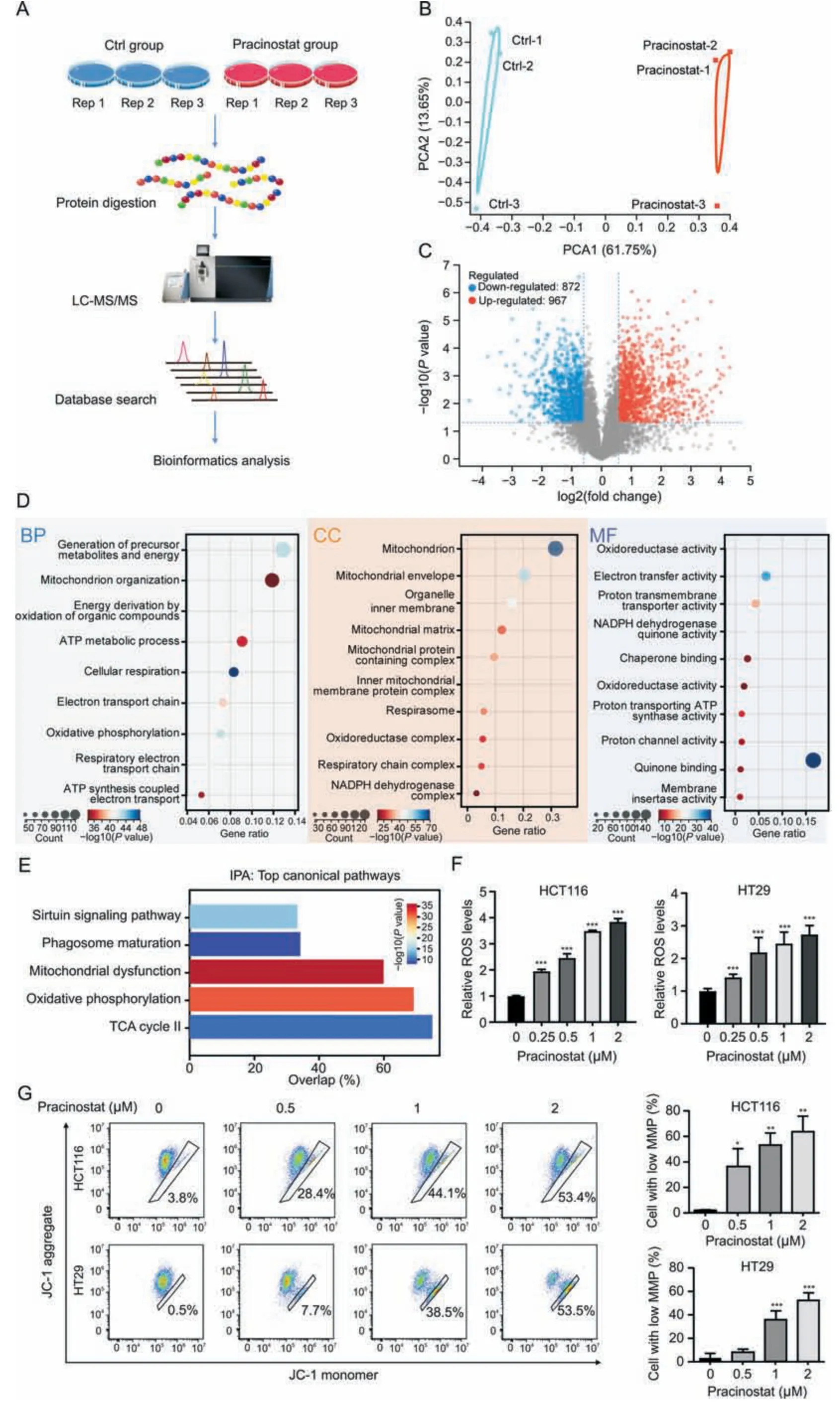
Fig.3.Pracinostat induces mitochondrial dysfunction.(A)The experimental workflow for the identification of protein alterations in colorectal cancer(CRC)induced by pracinostat.Lysates from HCT116 cells incubated with or without pracinostat (1 μM) were subjected to trypsin digestion and the peptides were subjected to mass spectrometry analysis.The proteomics result was performed in three biological triplicates (Rep1-3).(B) Principal component analysis (PCA) of identified proteins between Ctrl group and pracinostat treatment group.(C) Volcano map of differentially expressed proteins (DEPs;pracinostat vs Ctrl) identified by data independent acquisition (DIA)-based proteomics.(D) Gene ontology(GO)analysis of the DEPs regulated by pracinostat.The DEPs were enriched in terms of biological progression(BP),cellular components(CC),and molecular function(MF).(E)Ingenuity pathway analysis(IPA)of the DEPs regulated by pracinostat.(F,G)The effect of pracinostat on inducing colorectal cancer cells mitochondrial dysfunction.Both HCT116 and HT29 cells were treated with pracinostat (0-2 μM) as indicated for 24 h,and the cellular reactive oxygen species (ROS) was determined by flow cytometry using dichlorodihydro-fluorescein diacetate (F).The mitochondrial membrane potential (MMP) was determined by JC-1 assay,and the percentage of cell with low MMP was quantified (G).Data presented as Mean±standard deviation(SD)of three replicates;*P <0.05,**P <0.01,***P <0.001 when compared with Ctrl.Ctrl:control;LC-MS/MS:liquid chromatographytandem mass spectrometry;TCA: Trichloroacetic acid;JC-1: 5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide.
2.14.Serum biochemical test
The blood samples were placed at room temperature for 2 h and then separated at 2-8°C with a centrifugation speed of 3000 r/min for 15 min.The resulting supernatant was collected and tested using alanine aminotransferase (ALT) and aspartate aminotransferase (AST) detection kits (Huili,Changchun,China) according to the manufacturer's instructions.
2.15.Statistical analyses
All in vitro experiments were performed in three independent replicates,and all values are expressed as mean ± standard deviation.GraphPad Prism software (San Diego,CA,USA) was used to analyze the data.For multigroup comparisons,one-way ANOVA was conducted.The correlation between CDK5 and IC50 levels was evaluated using Spearman's correlation coefficient analysis.P<0.05 was considered statistically significant.
3.Results
3.1.Identification of pracinostat as an inducer of MFAD
Excessively asymmetric mitofission is inextricably linked to the process of programmed cell death [20],suggesting that pharmacologically inducting mitochondrial fission could provide novel insights into anticancer therapy.To identify novel MFAD inducers with anticancer activity against CRC,we screened 20 compounds from a commercially available pan-inhibitor library (Fig.S1 and Table S1)using the human CRC cell line HCT116 at a relatively low screening concentration (1 μM).Analysis of the mitochondrial morphology (stained with Mito-Tracker Red) in cells after treatment with individual compounds for 24 h revealed that pracinostat,a pan-HDAC inhibitor,significantly induced mitochondrial fragmentation in CRC cells,as evidenced by the elevated number of fragmented mitochondria,the decrease in tubular mitochondria and the ratio of rod/branch length (Fig.1A-C).More importantly,pracinostat (1 μM) exhibited the strongest anticancer effects on CRC compared to those of other compounds,with an inhibition ratio of 57% (Fig.1D).The effect of pracinostat on mitochondrial morphology was further confirmed by confocal microscopy;we found that elevated concentrations of pracinostat (0-2 μM)increased the number of fragmented mitochondria (Fig.1E).Moreover,TEM revealed that HCT116 cells treated with pracinostat displayed smaller and spherical mitochondria (Fig.1F).Examination of the mitochondrial dynamic markers demonstrated that pracinostat decreased the level of MFN1 and MFN2,while increased the phosphorylation of Ser616 of Drp1 (Fig.1G).Strikingly,Drp1 was significantly recruited to fragmented mitochondria after pracinostat treatment(Fig.1H).Collectively,our data demonstrate that pracinostat is an effective inducer of mitochondrial fission.
Next,we asked whether other HDAC inhibitors exhibit similar effect on inducing lethal mitofission.By employing SIS17 (HDAC11 inhibitor),CI994 (HDAC1/2/3 inhibitor),TMP195 (HDAC4/5/7/9 inhibitor),trichostatin A (TSA,pan-HDACi),and vorinostat (SAHA,pan-HDACi),we found that 1 μM of pracinostat,TSA,and SHAH showed stronger anticancer effects on HCT116 cells than that of SIS17,CI994,and TMP195 (Fig.S1B).Although pracinostat,SIS17,CI994 and TMP195 were able to increase the expression of FIS1 andp-Drp1,only pracinostat displayed both significant mitofission and strong anticancer effects(Fig.S1C).These results suggest that some HDAC inhibitors are able to induce lethal mitofission,while pracinostat is the promising one for cancer prevention.
3.2.Pracinostat exhibits significant anticancer effect on CRC
To investigate the anticancer effect of pracinostat on CRC,we performed the CCK-8 assay and observed that pracinostat decreased the cell viability of HCT116 and HT29 cells in dose-and time-dependent manners (Fig.2A),with IC50values of 0.44 ± 0.06 μM (HCT116) and 0.68 ± 0.05 μM (HT29) at 48 h,respectively.Long-term exposure to pracinostat led to decrease in the colony numbers of both HCT116 and HT29 cells,along with increasing concentrations of pracinostat (Fig.2B).Pracinostat also increased the percentage of Annexin V-positive cells,as well as the uptake of PI (Fig.2C).Moreover,cleaved forms of caspase 3 and PARP were elevated in both HCT116 and HT29 cells in line with increasing treatment of pracinostat (Fig.2D),suggesting that pracinostat suppresses CRC via apoptosis.To confirm the role of Drp1-mediated fission in pracinostat-induced apoptosis,we employed mitochondrial division inhibitor-1 (Mdivi-1),an effective inhibitor of Drp1,and found that the cleaved-PARP and cleaved-caspase 3 induced by pracinostat were blocked by pretreatment with Mdivi-1(5 μM,Fig.2E).An Annexin V/PI assay demonstrated that the percentage of early and late apoptotic cells (Annexin V positive)induced by pracinostat was decreased from~35% to~10% in both HCT116 and HT29 cells after Mdivi-1 treatment (Fig.2F).These results suggest that pracinostat induces cell apoptosis in a Drp1-dependent manner.
3.3.Pracinostat suppresses CRC by inducing mitochondrial dysfunction
We further investigated the mechanism underlying CRC inhibition by pracinostat.As shown in Fig.3A,DIA-based quantitative proteomics was performed on HCT116 cells treated with or without pracinostat.The identified protein numbers in pracinostat and control groups from triplicate experiments are shown in Fig.S2A,and show an equally normal distribution (Fig.S2B).Principal component analysis was performed to validate the reproducibility of each set of triplicate experiments (Fig.3B).A total of 5,704 proteins were co-identified in three experiments,with 967 proteins upregulated and 872 proteins downregulated in HCT116 cells treated with pracinostat (fold change ≥1.5,P<0.05;Fig.3C).GO analysis indicated that pracinostat-regulated DEPs were mainly involved in the biological processes of energy metabolism and mitochondrial organization.Most DEPs were enriched in the cellular components of mitochondria,the mitochondrial envelope,organelle inner membrane and mitochondrial matrix.In terms of molecular function,the pracinostat-regulated DEPs were mainly relevant to oxidoreductase activity and electron transfer activity(Fig.3D).These results were supported by IPA analysis,showing that the canonical pathways regulated by pracinostat included mitochondrial dysfunction and oxidative phosphorylation(Fig.3E).To confirm this effect,we detected the cellular ROS level by using dichloro-dihydro-fluorescein diacetate (DCFH-DA) staining and found that ROS was increased in both HCT116 and HT29 cells along with pracinostat treatment(Fig.3F).In addition,the percentage of low MMP cells was increased from~3%to 53%in both CRC cell lines with pracinostat stimuli (Fig.3G).Taken together,our DIA-based quantitative proteomics suggests that the anticancer effect of pracinostat on CRC is linked to mitochondrial dysfunction.

Fig.4.Cyclin-dependent kinase 5 (CDK5) is required for the anticancer effect of pracinostat on colorectal cancer cells.(A) Venn diagram depicting the intersection between data independent acquisition(DIA)-based proteomic result and dynamin-related protein 1(Drp1)-S616 kinase-prediction by iGPS software(http://igps.biocuckoo.org/index.php).A total of 8 potential kinases for Drp1-S616 were predicted.(B)Heatmap showing the protein expression level of the 8 potential kinases determined from the DIA-based proteomic results.Differentially expressed proteins were labeled with an asterisk.*P <0.05,**P <0.01.(C) Immunoblot analysis of the expression of CDK5 in HCT116 cells treated with elevating concentrations of pracinostat(0-1 μM).(D)Immunoblot analysis of CDK5 expression in HCT116 and HCT116-CDK5 cells treated with pracinostat as indicated.(E)The CCK-8 assay compared the cell viability of HCT116 cells and HCT116-CDK5 cells treated with increasing concentrations of pracinostat.(F) Colony formation assay of HCT116 cells and HCT116-CDK5 cells treated with or without pracinostat.(G) HCT116 cells and HCT116-CDK5 cells treated with or without roscovitine (1 μM) were exposed to increasing concentrations of pracinostat.The cell viability was determined by the CCK-8 assay.(H)Colony formation assay of HCT116-CDK5 cells treated with roscovitine and/or pracinostat as indicated.(I-O)Antitumor activity of pracinostat in CRC cells with or without CDK5 overexpression in xenograft model.HCT116 and HCT116-CDK5 cells were subcutaneously injected into nude mice(n =6).Mice were treated with pracinostat(25 mg/kg orally)or control(corn oil)every two days.(I)Representative images of xenograft tumors with indicated treatment.(J,K)Quantitative analysis of the tumor volume(J)and tumor weight(K)with indicated treatment.(L)Representative images of Ki-67 staining in xenografts tumors.Histogram compared the expression levels of Ki-67.(M) Statistical diagram of body weight of the nude mice with indicated treatment.(N,O) The serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were quantified.Data presented as Mean ± standard deviation (SD);n =3;one-way ANOVA;ns: not significant;*P <0.05,**P <0.01,***P <0.001.
3.4.CDK5 is critical for the anticancer effect of pracinostat on CRC
To further investigate the mechanism of how pracinostat induces mitochondrial fission-mediated cell death,we performed analysis by integrating 13 predicted kinases of Drp1 with our DIAbased quantitative proteomics results.Out of 8 potential hits,CDK5,mitogen-activated protein kinase 10 (MAPK10),and MAPK3 were significantly altered after pracinostat treatment (Figs.4A and B).Among these proteins,CDK5 was the top upregulated protein induced by pracinostat in a dose-dependent manner (Fig.4C).Moreover,the effect was amplified in CRC cells overexpressing CDK5 (Fig.4D).
CDK5 is well recognized as an oncogene in tumorigenesis,and here it was found to be highly expressed in primary CRC compared to normal tissues according to The Cancer Genome Atlas (TCGA) and Clinical Proteomic Tumor Analysis Consortium (CPTAC) databases(Figs.S3A and B).We also observed that HCT116 cells overexpressing CDK5 were more sensitive to pracinostat (IC50: 0.077 ± 0.06 μM)compared to that of the control (IC50: 0.564 ± 0.034 μM) (Fig.4E).Long-term treatment of pracinostat remarkably decreased the colony formation of CRC cells expressing a high level of CDK5(Fig.4F).By measuring the IC50for the six CRC cell lines treated by pracinostat(Fig.S3C),we found that the IC50s negatively correlated with the protein expression level of CDK5(Fig.S3D).Roscovitine,an inhibitor of CDK5,restored the anticancer effect of pracinostat on CRC cells expressing CDK5,as evidenced by cell viability and colony formation assays (Figs.4G and H).Consistently,the presence of roscovitine blocked the pracinostat-induced mitochondrial fission and MMP loss in CDK5-positive cells(Figs.S4A and B).
We further performed in vivo study to investigate the role of CDK5 in the anticancer effect of pracinostat using mice models subcutaneously injected with drugs every two days.As shown in Fig.4I,CDK5 overexpression increased tumor proliferation,whereas the tumor volumes in the pracinostat and CDK5/pracinostat groups were smaller than those in the control groups.Notably,the tumor size in the CDK5/pracinostat treatment group was smaller than that in the pracinostat treatment group.This result was also supported by the tumor growth curve(Fig.4J)and tumor weight (Fig.4K),as well as the Ki-67 and TUNEL staining of the xenograft tumors (Figs.4L and Fig.S4C).We found no significant loss in body weight (Fig.4M) or alteration of AST and ALT in the serum of mice with the various treatments(Figs.4N and O).These results suggest that pracinostat exhibits strong anticancer effect on CRC bearing high expression level of CDK5.
3.5.Pracinostat-induced CDK5/Drp1 signaling is required for peripheral fission of mitochondria
To validate whether CDK5 amplifies pracinostat-induced mitochondrial fission,we compared the mitochondrial morphology of HCT116 and HCT116-CDK5 cells treated with or without pracinostat by staining with Mito-Tracker.We found that pracinostat treatment significantly amplified the degree of mitochondrial fragmentation in HCT116-CDK5 cells,as compared to pracinostat treatment in HCT116 cells (Fig.5A).These effects were supported by the upregulation of fission markers,p-Drp1 and FIS1,and the decrease of fusion markers MFN1 and MFN2.Interestingly,another fission adaptor MFF was downregulated (Fig.5B).As two classical fission adaptors (FIS1 and MFF),their opposite expression patterns under pracinostat treatment or CDK5 expression indicate distinct scission signatures linked to mitochondrial fate[4].By using confocal assay,we observed that Drp1 was partially located in the midzone of mitochondria with CDK5 overexpression,as compared to diffuse Drp1 throughout the cytosol in untreated cells (Fig.5C).In CDK5-expressing cells,pracinostat treatment significantly induced the recruitment of Drp1 to mitochondria (Fig.5C),showing a fragmented mitochondria enriched with Drp1.Moreover,pracinostat stimulation dramatically induced the MMP loss in CRC cells with a high expression level of CDK5(Fig.5D).These findings suggest that pracinostat induces stronger Drp1-mediated asymmetric mitofission and results in mitochondrial dysfunction in CDK5-high CRC cells.
According to our kinase prediction of Drp1,as well as previous report [21],we further determined if there was a direct phosphorylation effect of CDK5 on Drp1-S616 by using a co-IP assay.As shown in Fig.6A,strong interactions between CDK5 and Drp1 were observed;moreover,the interaction was elevated by pracinostat in a dose-dependent manner and resulted in increased ofp-Drp1 levels (Fig.6B).A recent study revealed that scission occurring at the periphery of mitochondria is destined for mitochondrial damage(Drp1-FIS1 interaction),whereas division at the midzone leads to the proliferation of mitochondria(Drp1-MFF interaction)[4].We observed that Drp1 strongly interacted with FIS1 in HCT116-CDK5 cells treated with pracinostat,whereas the interaction between Drp1 and MFF was decreased in both the pracinostat and pracinostat/CDK5 groups,except for a slight increase in the CDK5 overexpression group (Fig.6C).To confirm this effect,the overall shape of the mitochondria was determined by TEM.More midzone fissions of mitochondria were observed in HCT116-CDK5 cells,and an elevated number of peripheral daughter mitochondria was found in the pracinostat/CDK5 group when compared to pracinostat alone (Fig.6D).ROS accumulation is another indicator of mitochondrial dysfunction.We determined mitochondrial ROS levels using MitoSOX staining in HCT116 cells.After 30 min of pracinostat exposure,the fluorescence intensity of ROS was elevated in peripheral daughter mitochondria (pracinostat and pracinostat/CDK5 groups) compared to that in non-dividing mitochondria (NC group) or daughters from midzone fissions(CDK5 group) (Fig.6E).Taken together,our results suggest that pracinostat induces CDK5-Drp1-FIS1 signaling activation to promote peripheral fission of mitochondria.
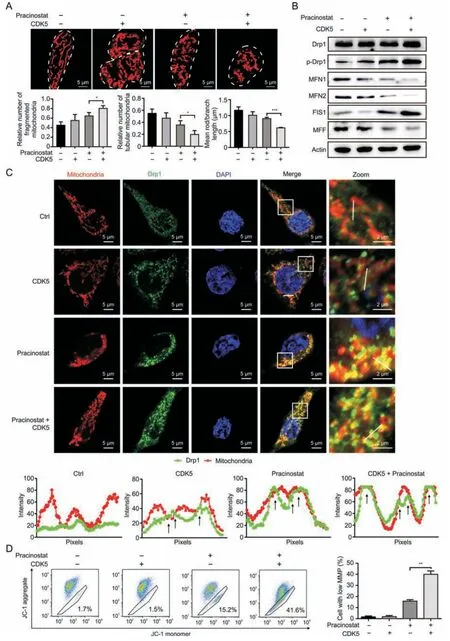
Fig.5.Pracinostat induces stronger dynamin-related protein 1 (Drp1)-mediated mitofission and mitochondrial dysfunction in cyclin-dependent kinase 5 (CDK5)-high colorectal cancer cells.(A) The mitochondrial morphology of HCT116 and HCT116-CDK5 cells with the indicated treatments was stained by Mito-Tracker-Red.The relative number of fragmented mitochondria,tubular mitochondria,and mean rod/branch length were statistically plotted,respectively.(B)Immunoblot analysis of the mitochondrial dynamics markers(p-Drp1 Drp1,MFN1,MFN2,FIS1 and MFF)in HCT116 cells and HCT116-CDK5 cells with indicated treatment.(C)Colocalization of Drp1 and mitochondria in CRC cells.HCT116 and HCT116-CDK5 cells treated with Ctrl(DMSO)or pracinostat were stained with Mito-Tracker-Red and subjected to microscopic analysis.Images in the box at higher magnification are shown in the right panels.The intensity profiles of Drp1 (green) and mitochondria (red) along the white line are plotted,with sites of colocalization marked by black arrows.(D)Mitochondrial membrane potential (MMP) of HCT116 and HCT116-CDK5 cells with indicated treatment was detected by JC-1.The percentages of cells with low mitochondrial membrane potential were quantified.Representative results were obtained from three independent experiments;error bars;standard deviation (SD).P values were indicated by unpaired Student's t-test.*P <0.05,**P <0.01,***P <0.001.MFN1:mitofusin 1;MFN2:mitofusin 2;FIS1:mitochondrial fission 1 protein;MFF:mitochondrial fission factor;DMSO:dimethyl sulfoxide;JC-1: 5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide.
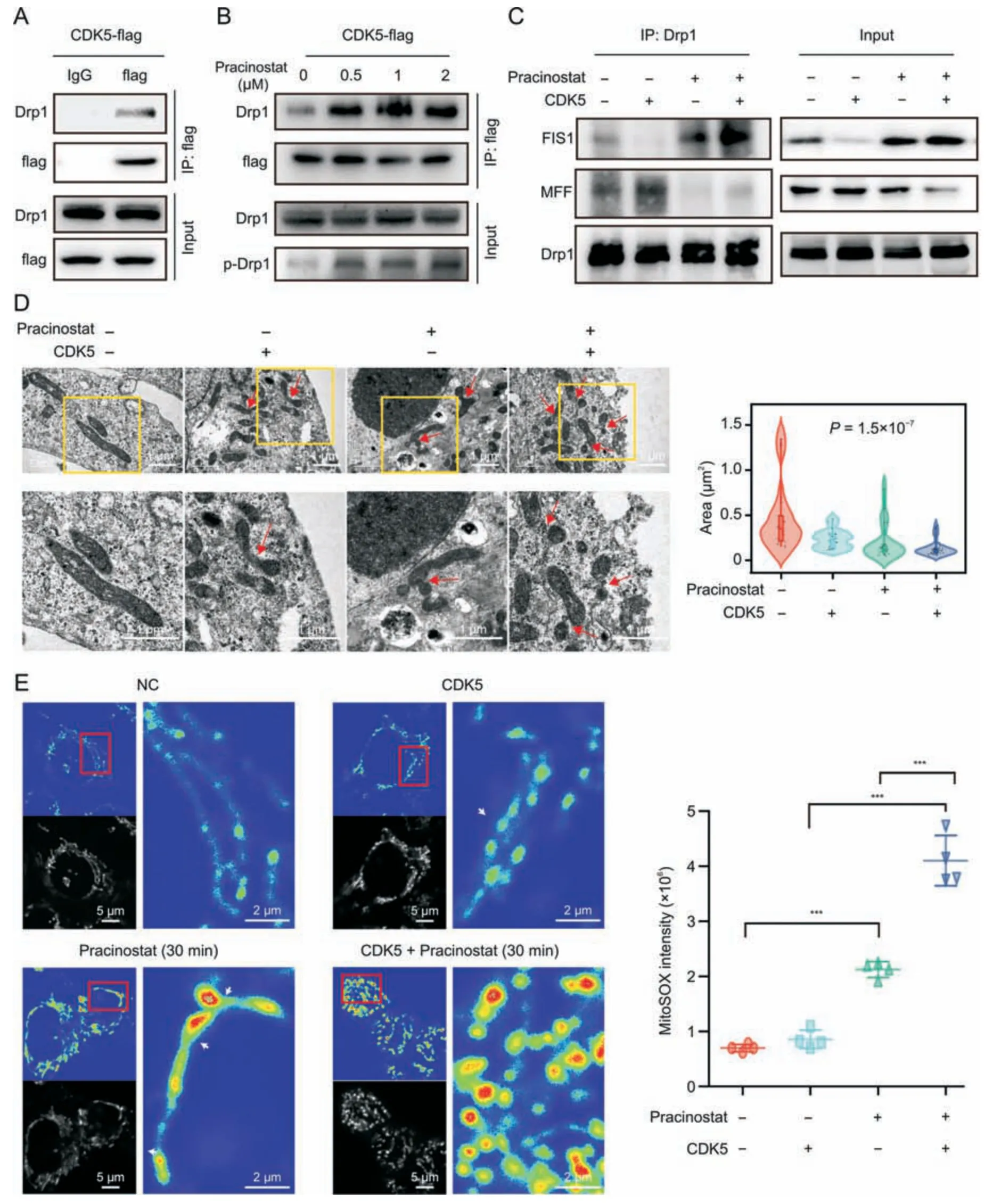
Fig.6.Pracinostat induces peripheral fission of mitochondria by activating cyclin-dependent kinase 5 (CDK5)/dynamin-related protein 1 (Drp1)/mitochondrial fission 1 protein(FIS1) signaling.(A) Co-immunoprecipitation (Co-IP) assay was performed to determine the interaction between CDK5 and p-Drp1.HCT116 stably expressing CDK5-flag was coimmunoprecipitated by anti-flag antibody.Expression of Drp1 and p-Drp1 was determined in CDK5 immunoprecipitates by Western blotting.(B) The interaction of CDK5 and p-Drp1 was detected in HCT116-CDK5 cells treated with increasing concentrations of pracinostat.(C)The interactions of Drp1 with FIS1 and mitochondrial fission factor(MFF)were detected in HCT116 cells with the indicated treatment.(D) The mitochondrial morphology of HCT116 cells with indicated treatment was evaluated using transmission electron microscopy.The area of mitochondria was quantified.n=15;nonparametric test.(E)MitoSOX labeling showing the mitochondrial reactive oxygen species(ROS)levels in peripheral or midzone positions of mitochondria with indicated treatment.The intensity of MitoSOX from 4 fields was quantified.The peripheral scission sites of mitochondria were marked by white arrows.Data presented as mean ± standard deviation (SD). P values were indicated by one-way ANOVA.***P <0.001.
3.6.Acetylation of lysine 33 is critical for CDK5 activation induced by pracinostat
Our proteomic results revealed that pracinostat increased the expression level of CDK5 in CRC.However,whether pracinostat influences the activity of CDK5 is unclear.Since pracinostat is a pan-HDAC inhibitor [22],we investigated whether pracinostat influences CDK5 activity by improving its acetylation level.Our co-IP assay demonstrated that increasing concentrations of pracinostat(0-2 μM)elevated the K-acetylation of CDK5,accompanied by the increased affinity to CDK5R1 (Fig.7A).This suggests that pracinostat regulates CDK5 activity through promoting its acetylation.According to the Uniprot database,CDK5 contains two conserved acetylation residues,lysine 33 (K33) and lysine 56 (K56) (Fig.7B).To compare their importance in the anticancer effect of pracinostat,we generated arginine substitution of K33 and K56 mutations(K33R and K56R) in CDK5,respectively,and found that cells with K33R,but not K56R,in CDK5 showed resistance to pracinostat,as compared to CDK5-wild type (WT) (Fig.7C).Indeed,K33R displayed a lower interaction with CDK5R1,even in the presence of pracinostat (Figs.7D and E),suggesting that acetylation of K33 in CDK5 is critical for its activation upon pracinostat stimulation.Long-term treatment with pracinostat resulted in a higher colony formation ability of CDK5-K33R cells compared to that of CDK5-WT cells in response to pracinostat(Fig.7F).Furthermore,as compared to the fragmented mitochondria in CDK5/pracinostat cells,HCT116 cells with CDK5-K33R showed a more tubular mitochondrial morphology,with increased mitochondrial tubule length in the presence of pracinostat(Fig.7G).Moreover,the loss of MMP in HCT116 cells expressing CDK5-K33R was smaller than that in CDK5-WT in response to pracinostat (Fig.7H).
Finally,we determined the effect of CDK5-K33 acetylation on the anticancer activity of pracinostat in vivo.By using the xenograft model,we found that HCT116-CDK5 cells were sensitive to pracinostat,while the CDK5-K33R group was refractory to pracinostat treatment.This observation was supported by the tumor growth curve (Figs.7I and J),tumor weight (Fig.7K),Ki-67,staining and TUNEL staining of xenograft tumors (Figs.S5A and B).We did not find a significant loss of body weight (Fig.S5C).These results suggest that pracinostat induces MFAD in CRC by promoting K33 acetylation of CDK5.
4.Discussion
CRC accounts for approximately 10% of all annually diagnosed cancers and cancer-related deaths worldwide[23].CRC incidence is predicted to increase to 2.5 million new cases in the next decade[24,25].In precision medicine,identifying specific biomarkers and understanding the related mechanisms will lead to more effective therapeutic strategies for cancer treatment.We recently demonstrated that targeting acid sphingomyelinase by polyphyllin D significantly improves the anticancer effect of sorafenib on hepatocellular carcinoma [26].This current work revealed that the HDACi pracinostat was a novel inducer of mitofission-associated cell death and exhibited a significant anticancer effect on CRC cells via CDK5,a previously identified oncogene,at both the protein and post-translational levels.Decrease of CDK5 activity caused resistance to pracinostat.Mechanistically,we identified the CDK5-Drp1-FIS1 axis as the central signaling pathway for upregulating mitochondrial peripheral fission in CRCs upon pracinostat stimuli,resulting in excessive mitofission and mitochondrial apoptosis.
Abnormal expression of CDK5 is associated with malignant tumor initiation and progression[27].In colon cancer,CDK5 directly phosphorylates extracellular-signal-regulated kinase 5 (ERK5) at Thr732 to promote the progression of CRC carcinogenesis[14].The kinase activity of CDK5 highly relies on non-cyclin activators CDK5R1 and CDK5R2 [28];more importantly,this process is modulated by post-translational modifications [13].K33 and K56 are two conserved acetylation residues of CDK5,whose acetylation status is controlled by lysine acetyltransferase 2 A and sirtuin 1[29].In this study,we found that CDK5 was regulated by pracinostat at both the protein and PTM levels,as evidenced by increased CDK5 protein and acetylation levels after pracinostat treatment.This effect can be explained by the inhibition of HDAC (pracinostat treatment),which increases histone acetylation and thus drives the expression of multiple genes.As for which HDACs regulates the acetylation of CDK5 warrants further investigation.
Notably,we discovered that pracinostat-mediated HDAC inhibition increased the acetylation of CDK5,and acetylated-K33 contributed more than acetylated-K56 to the anticancer effect of pracinostat on CRC.Acetylation of CDK5 displays higher kinase activity,whereas arginine substitution of K33 (K33R) reduces its activity,as evidenced by the decrease of the physiological interaction between CDK5 and CDK5R1.In neurons,prolonged activation of CDK5 acts as an apoptosis-enhancing signal that inactivates peroxiredoxin 2 (an antioxidant enzyme) and myocyte enhancer factor 2D (a survival promoting transcription factor),respectively[30,31].Herein,we measured the IC50 of pracinostat in several CRC cell lines with different levels of CDK5.Interestingly,a negative correlation was found between CDK5 expression level and the IC50(Fig.S3D).CRC cells with CDK5 overexpression bore fragmented mitochondria,displaying higher cell proliferation and tumor growth,while CRC cells with such an oncogenic phenotype were sensitized to pracinostat treatment,as compared with CRC cells expressing the CDK5-K33R mutation.This finding provides a new explanation regarding the controversial roles of CDK5 in apoptosis and cancer progression.We speculated that treatment with commercial CDK5 activators may enhance the anticancer effect of pracinostat in combined therapy.
The answer to why CRC cells with high level of CDK5 are sensitized to pracinostat can be traced back to the question why Drp1-mediated mitochondrial fission results in dual fates of mitochondria.Many kinases,such as ERK,AMP-activated protein kinase and AKT Serine/Threonine Kinase1,have been reported to phosphorylate Drp1 at S616 to induce mitochondrial division at the midzone,promoting metabolism reprogramming and biogenesis[32].Consistently,the fragmented mitochondria in CRC cells with high levels of CDK5 were the result of midzone fission driven by CDK5-Drp1 signaling,showing a low level of ROS and high MMP through the mitochondria.However,damaged mitochondria undergo mitochondrial peripheral fission destined for mitophagy or cell death.We found that pracinostat induced peripheral fission of mitochondria in CRC,with a high ROS and low MMP observed in positions at the periphery of mitochondria(0-25%from a tip).This effect could be explained by the fact that two classical fission adaptors MFF and FIS1 showed opposite expression trend after pracinostat treatment.The increased FIS1 and decreased MFF induced by pracinostat provided a fundamental for Drp1 to switch its binding partners from MFF to FIS1,resulting in the distinct fission signatures (peripheral or midzone).Notably,fragmented mitochondria in cancer cells are vulnerable in response to mitochondria-targeted agents.Nevertheless,the mechanism of how MFF and FIS1 display opposite expression after pracinostat treatment warrant further investigated.
In view of the potential application of pan-HDACi pracinostat in clinic,we found that other HDACi TSA and SAHA showed comparable anticancer effects on CRC with pracinostat,however,TSA and SHAH seemed to suppress CRC independent of FIS1 and Drp1[33],indicating that only part of HDAC inhibitors are able to induce MFAD.Mitochondrial scission at peripheral and midzone links to different cell fates.Pharmacological induction of excessively asymmetric MFAD represents a new therapeutic strategy for clinical cancer therapy.This study identified pracinostat as a novel MFAD inducer that selectively suppresses CDK5-high CRC.
5.Conclusions
In summary,our study establishes a proof-of-concept for distinct mitochondrial fission signatures in cancer in response to anticancer drugs.It also provides solid evidence to support the hypothesis that the pracinostat-regulated CDK5-Drp1-FIS1 signaling axis triggers mitochondrial peripheral scission and ultimately leads to excessive mitochondrial fission-associated apoptosis in CRC.Nevertheless,our current evidences do not prove whether CDK5 is the sole inducer of Drp1-FIS1 signaling.Understanding the precise role of CDK5 in CRC and the regulation of mitochondrial fission not only advance our knowledge of CRC pathogenesis but also allow for the development of potential pracinostat-based CRC therapeutic strategies.
CRediT author statement
Xiao-Ling Liang:Investigation,Writing -Original draft preparation,Visualization;Lan Ouyang:Investigation,Data curation,Methodology;Nan-Nan Yu:Investigation;Zheng-Hua Sun:Software,Methodology;Zi-Kang GuiandYu-Long Niu: Formal analysis and Validation;Qing-Yu He:Writing-Reviewing and Editing,Project administration;Jing Zhang:Conceptualization,Methodology,Project administration,Writing-Reviewing and Editing;Yang Wang:Conceptualization,Methodology,Project administration,Writing -Reviewing and Editing.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant Nos.: 82103208,and 82002948),the Guangdong Basic and Applied Basic Research Foundation (Grant Nos.: 2022A1515220212,and 2023A1515030115),National Key R&D Program of China (Grant No.: 2020YFE0202200) and Jinan University National College Students' Innovation and Entrepreneurship Training Program (Program No.: 202110559085).
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2023.06.005.
杂志排行

Journal of Pharmaceutical Analysis的其它文章
- Protective effects of catalpol on cardio-cerebrovascular diseases: A comprehensive review
- Recent advancements in single-cell metabolic analysis for pharmacological research
- SPME as a green sample-preparation technique for the monitoring of phytocannabinoids and endocannabinoids in complex matrices
- Monoclonal antibody targeting mu-opioid receptor attenuates morphine tolerance via enhancing morphine-induced receptor endocytosis
- Protective effects of dioscin against Parkinson's disease via regulating bile acid metabolism through remodeling gut microbiome/GLP-1 signaling
- Dihydroartemisinin ameliorates innate inflammatory response induced by Streptococcus suis-derived muramidase-released protein via inactivation of TLR4-dependent NF-κB signaling