光学活性薄荷醇异构体的合成、衍生及分离
2023-12-04戈光琼付延明朱成峰李有桂
戈光琼, 付延明, 沈 昊, 吴 祥, 朱成峰, 李有桂
(合肥工业大学 化学与化工学院,安徽 合肥 230009)
0 引 言
L-薄荷醇凭借其独特的清凉、杀菌和止痒等多种功效[1-3],在精细化学品、香精香料、医疗卫生和食品[4-7]等方面被广泛应用;由于薄荷醇自身的结构特性,还可以用作药物中间体[8]或者通过不对称催化反应[9]来制备相应的旋光性化合物等。
L-薄荷醇分子式中含有3个手性碳中心,对应着4对对映异构体和8个立体异构体[8-9],如图1所示。当薄荷醇六元环上的取代基所处平伏键和直立键的位置不同时,会导致每种异构体之间的能量存在差异,决定了每种立体异构体的特性,其中D/L-薄荷醇具有相对更明显的清凉功效,这也使其具有更高的工业价值[4-7]。

图1 薄荷醇8种立体异构体的结构
对于薄荷醇的合成路线,已有学者进行了研究。文献[10]报道以ZnBr2/SiO2为催化剂,以香茅醛为原料制备L-薄荷醇,但获得的产物几乎为异胡薄荷醇;文献[11]报道了在β-环糊精存在下,使用Rh-Al2O3催化剂催化百里酚加氢(转化率接近95%),对D/L-薄荷醇具有中等的选择性;文献[12]报道了一种实用的Co-PMA-PZ@SiO2-800 MOF催化剂,在150 ℃、50 bar的氢气氛围中,百里酚转化率可以达到92%,并对D/L-薄荷醇具有较好的选择性。
以上研究表明,现有方法合成的薄荷醇大多为外消旋体,而通过不对称催化来选择性地合成单一构型的薄荷醇难度较大,且使用的催化剂价格昂贵并难以合成[13-14]。基于此,本文通过手性液相的方式来研究薄荷醇色氨酸酯的分离情况,并建立一种相对高效地分离和鉴定薄荷醇光学异构体的方法。
在药学和生理学研究领域,手性化合物已引起人们的普遍兴趣[15-16],而高效液相色谱手性固定相法(high performance liquid chromatography-chiral stationary phase,HPLC-CSP)在对映体化合物的分离分析和制备方面表现出独特的优势[17]。手性固定相(chiral stationary phase,CSP)是整个高效液相色谱(high performance liquid chromatography,HPLC)系统的关键部件,直接决定了分离的效果,使用具有良好手性识别能力的手性固定相进行直接分离,可作为一种既适用于制备又适用于分析的简单实用方法,并已经得到了显著发展[18-20]。
目前,基于半合成大分子中特有的各种不同分子和超分子结构特点开发出的多糖类手性固定相的适用样品类型范围十分广泛,这使得利用合适的手性固定相以及流动相进行对映体的拆分往往具有高效的结果[21]。
本文采用单一构型的手性氨基酸[22]对单一构型对映体的薄荷醇进行酯化衍生化反应,不仅有助于增强薄荷醇在紫外波长下的吸收从而极大提高紫外的检测效果,同时衍生出的酯也具有很好的化学和光学稳定性。经过手性氨基酸衍生后的薄荷酯也是具有立体结构差异的多手性中心非对映异构体,根据非对应异构体的空间结构以及性质的差异[23],在HPLC-CSP系统中确定合适的分离参数以达到合适的分离效果,从而能够建立一种针对经手性氨基酸衍生后的薄荷醇的分离和检测方法。
1 实验部分
1.1 仪器和试剂
所有HPLC实验均使用配备有Agilent 1260 Infinity标准自动进样器、Agilent 1260 Infinity柱温箱、Agilent 1260 Infinity VL型四元泵和Agilent 1260 Infinity VWD可变波长检测器的Agilent 1260 HPLC仪器(Agilent Technologies, Santa Clara, USA)进行操作;Workstation软件Agilent OpenLAB CDS DESKTOP-TDDMRJ7用于仪器控制,数据采集和数据处理软件为Agilent OpenLAB Data Analysis(版本2.203.0573)。分离系统中手性柱为新型键合型淀粉-三(3,5-二甲基苯基氨基甲酸酯)手性色谱柱(Chiralpak IA)、新型键合型纤维素-三(3,5-二氯苯基氨基甲酸酯)手性色谱柱(Chiralpak IC)、硅胶表面涂敷型直链淀粉-三(3,5-二甲基苯基氨基甲酸酯)手性色谱柱(Chiralpak AD)和硅胶表面涂敷型纤维素-三(3,5-二甲基苯基氨基甲酸酯)手性色谱柱(Chiralpak OD);正相系统的HPLC分离是在20 ℃下以不同体积比的异丙醇、正己烷混合溶剂作为流动相,并以一定的流动相流速进行,紫外检测在波长为254 nm下进行。
本文使用的起始原料D/L-薄荷醇购于安耐吉公司,反应中使用的试剂环己烷、无水硫酸镁、4-二甲氨基吡啶、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、二碳酸二叔丁酯等均购于上海阿拉丁生化科技股份有限公司;L-色氨酸、重铬酸钾、硼氢化钠、盐酸、硫酸、反应溶剂四氢呋喃、甲醇以及HPLC级溶剂(如异丙醇和正己烷)等均购于国药集团化学试剂公司。
1.2 不同构型薄荷醇以及色氨酸酯的合成
8种薄荷醇异构体的合成线路如图2所示。首先,分别从已有的单一构型的D/L-薄荷醇(3.12 g,20 mmol)出发,通过氧化合成得到单一构型的孟酮(3.08 g,99.5%),再与吡咯经由烯胺中间体水解后转化为2种不同构型的孟酮(0.65 g,21.0%),由此得到4种构型不同的孟酮;进一步将其还原,并通过柱层析进行分离,最终可以合成出其余构型的薄荷醇(新薄荷醇0.74 g,24.1%;新异薄荷醇0.15 g,4.56%)。由于D/L-异薄荷醇的结构不够稳定而未得到,其余每种醇和酮的化学结构均由1H NMR和13C NMR表征,且与文献[24]中的结构一致。8种非对应异构体薄荷醇色氨酸酯的合成线路如图3所示。

图2 8种薄荷醇异构体的合成线路
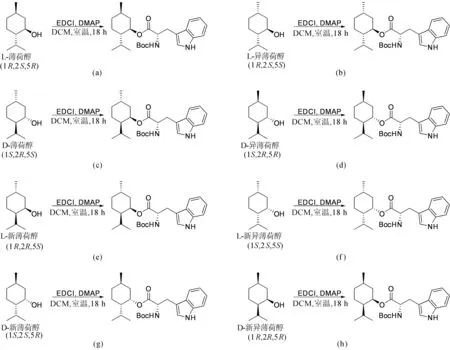
图3 8种非对应异构体薄荷醇色氨酸酯的合成线路
将已得到的不同构型的薄荷醇(0.78 g,5 mmol),在EDCI(1.05 g,5.5 mmol)和DMAP(0.061 g,0.5 mmol)的作用下分别与Boc-L-色氨酸(1.52 g,5 mmol)发生缩合反应,从而得到非对应异构体薄荷醇色氨酸酯(2.19 g,99.1%),每种酯的化学结构由1H NMR和高分辨率质谱进行表征确认。
2 结果与讨论
2.1 手性分离机制
直接手性分离的机制是通过手性固定相与作为分析物的对映体之间发生相互作用,从而形成瞬时非对映体复合物而实现分离。复合物的形成是氢键、偶极-偶极相互作用(定向力)、偶极-诱导偶极(诱导力)、诱导偶极-瞬时偶极(分散力)、π-π相互作用、静电相互作用和包容复合的结果[25]。
分析物的分离效果直接由手性固定相决定,对映体在手性柱上的分离依靠溶质和手性固定相上的极性氨基甲酸酯基团之间的相互作用,而后者则通过利用手性固定相和薄荷醇色氨酸酯中的C=O和N—H基团的氢键与溶质发生相互作用。此外,手性固定相上的C=O基团和薄荷醇色氨酸酯上的C=O基团之间还会发生偶极-偶极作用。
薄荷醇色氨酸酯和螺旋直链淀粉衍生物同时具有多个手性中心,因此聚合物所含有的大量手性活性位点与溶质发生相互作用的概率较高,可产生不同的相互作用效果,最终达到将非对映体分离的效果。
2.2 薄荷醇色氨酸酯的分离条件
2.2.1 手性柱的筛选
将得到的8种经过结构表征的酯分别使用Chiralpak IA、IC、AD、OD色谱柱(250 mm×4.6 mm×5 μm),在HPLC-CSP中对分析物进行分离和检测。流动相为异丙醇、正己烷,两者体积比为1∶9,流速为1.0 mL/min,检测波长为254 nm,柱温设定为20 ℃。混合酯样品是将8种构型酯混合均匀后再进行检测,检测时每种构型酯的样品注入量为10~20 μL。
将混合后的酯和单一构型的酯分别在HPLC-CSP上进行分离和检测,通过比对Chiralpak IC柱中混合酯和单一构型酯的出峰保留时间确定分离效果,如图4所示。
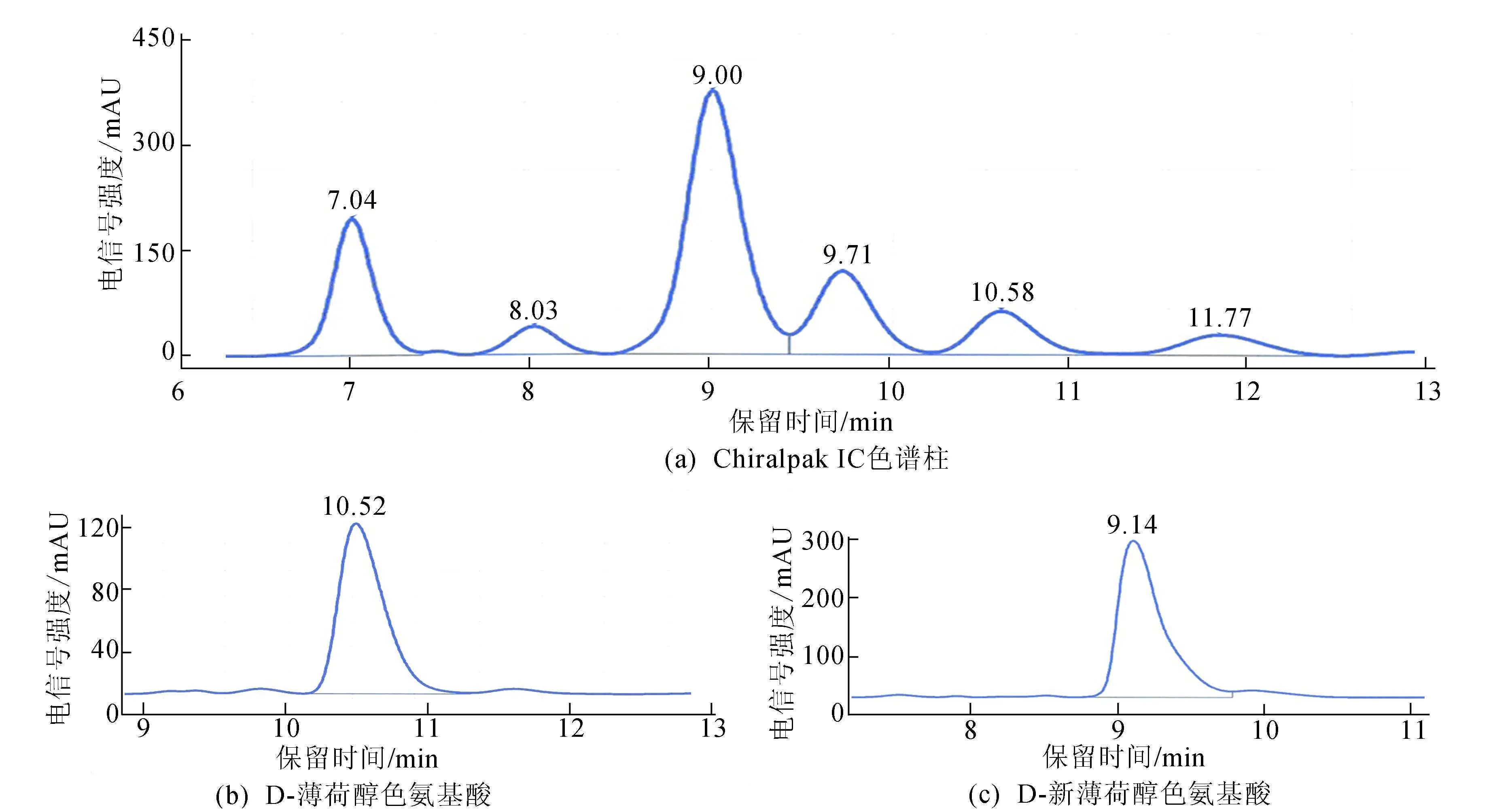
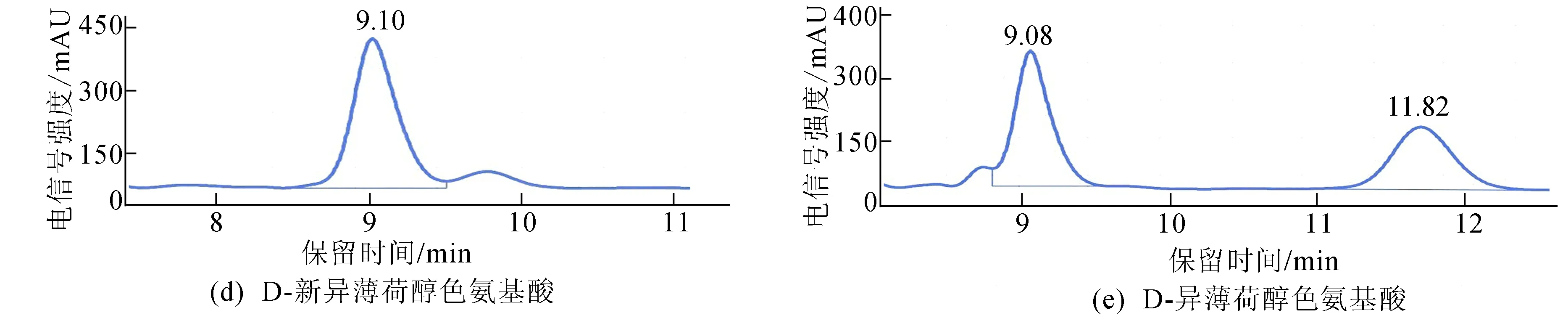
由图4可知,8种构型酯的出峰顺序依次为L-薄荷醇色氨酸酯、L-新薄荷醇色氨酸酯、D-新异薄荷醇色氨酸酯、D-新薄荷醇色氨酸酯、L-新异薄荷醇色氨酸酯、D-薄荷醇色氨酸酯、L-异薄荷醇色氨酸酯和D-异薄荷醇色氨酸酯,每个色谱峰的分离度R(resolution)分别为1.556、1.391、0.898、1.143、1.608。但D-新异薄荷醇色氨酸酯、D-新薄荷醇色氨酸酯、D-薄荷醇色氨酸酯以及L-异薄荷醇色氨酸酯这4种混合物之间无法有效完全地完全分离开来。
本文利用Chiralpak IA柱分别对混合酯和8种单一构型酯进行分离并比对出峰时间,如图5所示。

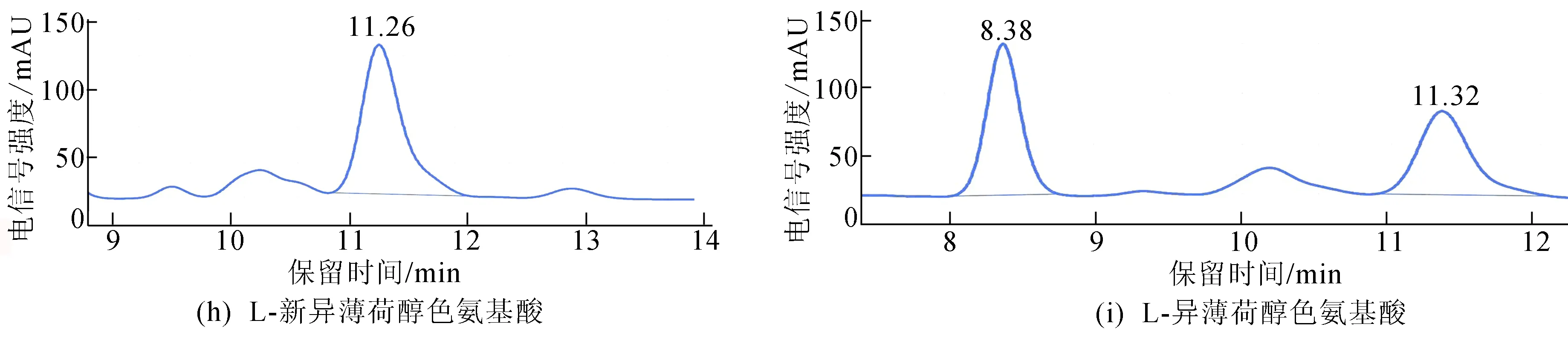
图5 利用Chiralpak IA柱分离8种混合酯和检测单一构型的酯
从图5可以看出:峰顺序依次为L-异薄荷醇色氨酸酯、D-薄荷醇色氨酸酯、L-新薄荷醇色氨酸酯、D-新薄荷醇色氨酸酯、L-新异薄荷醇色氨酸酯、D-新异薄荷醇色氨酸酯、L-薄荷醇色氨酸酯、D-异薄荷醇色氨酸酯,且每个色谱峰的分离度R分别为1.481、0.936、1.778、0.456、1.521;在液相图谱上显示出了6个峰,L-薄荷醇色氨酸酯和D-异薄荷醇色氨酸酯在液相图谱上以单一峰呈现;在此条件下,D-薄荷醇色氨酸酯和L-异薄荷醇色氨酸酯同样也不具有较好的分离效果,这可能是由于这两对非对映体仅在薄荷醇六元环五号位甲基位点的构型上存在差异,从而使得手性填充物质纤维素-三(3,5-二甲基苯基氨基甲酸酯)与非对映酯的手性位点之间的相互作用力太接近,从而在手性填充柱上无法实现完全分离。
2.2.2 流动相极性和流速的筛选
在筛选了初步的分离条件后,可通过调节流动相的极性大小和流速快慢来达到最佳的分离效果。实际上,流动相决定了分析物和选择剂互动位点的溶剂化程度以及这些位点能否用于分析物与手性固定相之间的分子接触,因此,流动相在对映选择中扮演着重要角色,选择合适的流动相也是高效分离方法获取的重要环节[21]。此外,流速的变化也能在一定程度上影响上述参数。
本文采用变换流动相极性和流速的方式(由大到小梯度分离;考虑到程序的设定终止时间,筛选时的最小流速为0.5 mL/min),先后采用了异丙醇与正己烷体积比为1∶9、8∶92、7∶93、5∶95的流动相组成以及1.0 、0.8 、0. 5 mL/min的流速比较分离效果,根据筛选分析不同流动相组成以及流速的分离效果发现,流动相的极性越小,流动的流速越慢,混合薄荷醇色氨酸酯的分离效果就越好。
在Chiralpak IA柱中,当将流动相异丙醇与正己烷体积比5∶95、流速为0.5 mL/min时,可以发现液相谱图中的各色谱峰之间具有较好的分离效果;当使用Chiralpak IC柱时,流动相异丙醇与正己烷体积比为1∶9、流速为0.8 mL/min时,液相谱图中的各色谱峰之间具有较好的分离效果。
3 结 论
本文研究了经过手性氨基酸衍生化后的薄荷醇在HPLC-CSP上的分离效果,并通过使用不同的手性填充柱、改变流动相的组成和流速来改善分离效果。使用Chiralpak IA柱分离时,流动相异丙醇与正己烷体积比5∶95、流速为0.5 mL/min时,可以取得较好的分离效果;当使用Chiralpak IC柱时,流动相异丙醇与正己烷体积比为1∶9、流速为0.8 mL/min时可达到较好的分离效果。基于本实验的结果分析,在接下来工作中可通过改变手性衍生试剂进一步实现完全分离薄荷醇异构体的目标。
