基于WGCNA分析及风险评分构建头颈部鳞状细胞癌的预后模型
2023-12-01金祥
金祥
头颈部鳞状细胞癌(head and neck squamous cell carcinoma,HNSCC)多发于口腔、鼻、咽、喉的黏膜上皮细胞,是全世界第六大常见的恶性肿瘤[1]。危险因素包括吸烟、酗酒、环境致癌物质及HPV感染等。大多数HNSCC的患者发现确诊时已处于晚期[2]。尽管手术技术、放化疗及免疫治疗的应用显示良好的效果,但五年生存率仅有50%[3]。其中一个重要的原因是缺乏针对早期HNSCC的有效生物标志物的认识与应用以便患者及时医治。近年来,靶向治疗已多应用于临床治疗HNSCC一线方案[4-5],因此,寻找HNSCC的预后标志物对于为患者提供有效的诊疗方案及延长患者的生存时间存在迫切而又深远的意义。在本研究中,使用R软件整合了GEO(gene expression omnibus)[6]和ArrayExpress数据库[7]的芯片数据,用TCGA(the cancer genome atlas)[8]的样本数据确定了11 个与HNSCC的生存有关的候选基因,使用LASSO回归分析及列线图构建了预测患者生存率的预后模型。
1 资料与方法
研究方法及技术路线如图1。
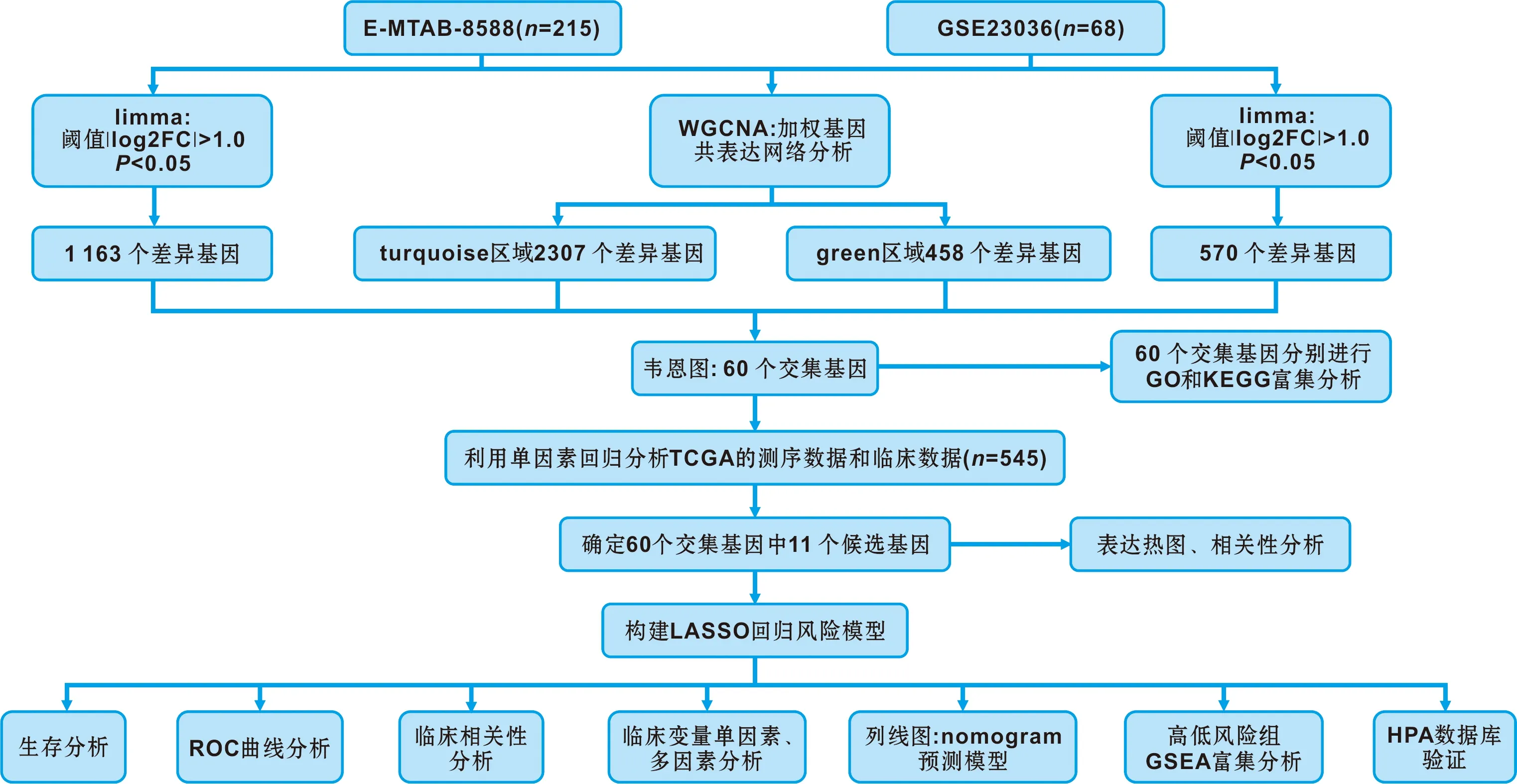
图1 预后模型构建的流程图
1.1 数据准备
68 个HNSCC样本(GSE23036)[9]的芯片数据从GEO (https://www.ncbi.nlm.nih.gov/geo/) 数据库下载,其中包括63 个癌症样本及5 个正常组织样本。E-MTAB-8588队列的芯片数据包括117 个癌症样本及98 个正常组织样本从ArrayExpress (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-8588/) 数据库中获取。 545 个样本的测序数据及临床数据从TCGA (https://portal.gdc.cancer.gov/) 数据库获取。
1.2 获取GEO和ArrayExpress数据集的交集基因
以校准后的P值<0.05及|log2FC|>1.0为阈值,使用R语言的limma包对GSE23036和E-MTAB-8588校正后的数据进行差异分析。利用加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)包[10]进行加权基因共表达网络分析,得到两个队列P<0.05的模块。选取两个队列的差异基因及P值最小的模块基因取交集,利用韦恩图[11]进行可视化。得到的交集基因GO和KEGG通路富集分析,GO分为3 个部分:生物过程(BP)、细胞组成(CC)和分子功能(MF)。
1.3 在TCGA队列中建立预后模型
首先利用单因素Cox分析确定交集基因中与HNSCC生存相关的基因。利用R包glmnet进行LASSO回归分析建立风险模型,每个患者的风险值运用以下公式计算:
Coefi表示模型中基因的回归系数,Expi表示基因的表达量。以中位风险值为标准将患者分为高低风险两组。
1.4 基因预后模型的验证
使用Kaplan-Meier (KM)生存分析比较高低风险两组患者的生存率,绘制操作者工作特征(ROC)曲线,利用曲线下面积(AUC)验证模型的预测能力。随后,利用单因素及多因素Cox回归分析验证风险评分和临床变量(性别、年龄、肿瘤分级、分期及TN分期)的预后独立性,利用R包rms建立列线图预测患者生存率。在高低风险组中使用GSEA软件(4.1.0版本)进行基因集富集分析。在HPA(Human Protein Atlas,https://www.proteinatlas.org/)数据库中验证模型中的基因表达水平。
1.5 统计学方法
使用PERL软件(ActivePerl- 5.28.msi,http://www.perl.org) 及R软件(4.1.1版本)进行所有的数据分析。
2 结 果
2.1 在GEO及ArrayExpress中筛选出交集基因
以|log2FC|>1.0和FDR<0.05为阈值在E-MTAB-8588和GSE23036队列中分别筛选出1 163和570 个差异基因(图2A~B)。应用WGCNA筛选特异模块,在E-MTAB-8588和GSE23036队列中选取软阈值β=7和5,分别得到14和12 个模块(图2C~D);选取最有意义的模块,E-MTAB-8588中的turquoise及GSE23036的green模块,分别包括2 307及458 个基因,与差异基因取交集,得到60 个交集基因(图3A)。GO富集分析显示60 个基因在脂肪酸代谢途径、药物代谢过程、环氧合酶P450途径等生物过程中富集(图3B),KEGG显示基因在细胞色素P450代谢、化学致癌、花生四烯酸代谢等通路富集(图3C)。
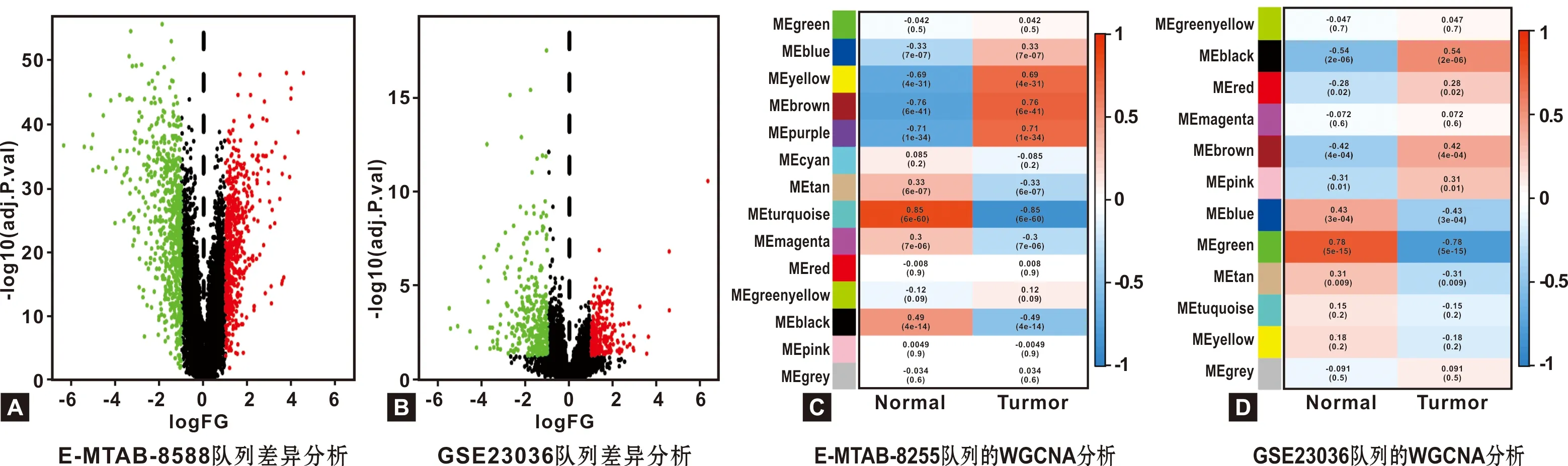
图2 基因差异分析及共表达网络分析
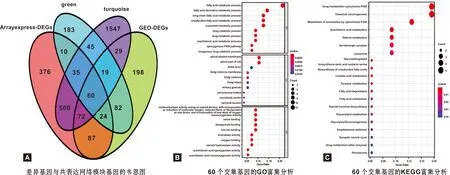
图3 交集基因及富集分析
2.2 在TCGA中建立预后模型
利用单因素回归分析确定60 个交集基因中有11 个基因(EXPH5,ELOVL6,ACOX3,NAGK,LPIN1,TJP3,NBEAL2,SASH1,MGST2,TSPAN6,ADH7)与HNSCC的生存有关(图4A);其中相关性分析(图4B)显示NBEAL2与EXPH5及SASH1显著正相关(R=0.66)。热图可视化11 个基因在TCGA正常及肿瘤样本中的表达量(图4C)。通过LASSO回归建立11基因的预后模型(图5A~B),利用基因回归系数及表达量计算风险值:riskscore=(-0.233)*EXPH5+(0.104)*ELOVL6+(-0.026)*ACOX3+(-0.036)*NAGK+(-0.197)*LPIN1+(-0.054)*TJP3+(-0.005)*NBEAL2+(0.014)*SASH1+(0.018)*MGST2+(0.022)*TSPAN6+(0.003)*ADH7基于中位风险值,498 例患者被分为高低风险两组。图5C显示风险值和生存状态的分布及预后基因随风险值的表达量。绘制热图评估高低风险组中预后基因表达量与临床变量的相关性(图6),T分期(P<0.001),临床分期(P<0.01)及N分期(P<0.05)在高低风险两组中有显著差异。
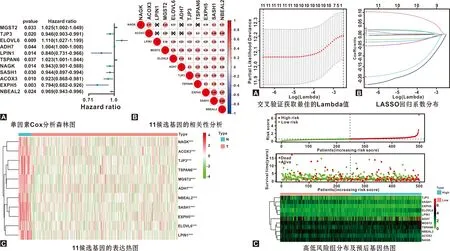
图4 预后相关基因分析 图5 构建LASSO回归风险模型
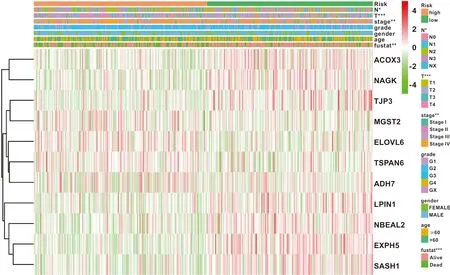
图6 临床变量与预后基因相关性的热图
2.3 验证模型的有效性
Kaplan-Meier生存分析显示高风险组患者预后不良(P=8.002e-07,图7A)。同时绘制ROC曲线,1、3、 5 年的AUC分别为0.67、 0.70、 0.66(图7B~7D),提示模型具有一定的预测效果。进行单因素及多因素Cox回归分析评估风险比(HR),95%可信区间(CI)及P值,单因素回归分析显示风险评分、临床分期、年龄和TN分期与患者生存有关(图7E);多因素分析结果显示风险评分(HR=1.784,95%CI:1.461-2.180,P<0.001),年龄(HR=1.023,95%CI:1.005-1.040,P<0.001)及N分期(HR=1.332,95%CI:1.054-1.683,P=0.016)具有独立预后性(图7F)。随后,选择上述3 个具有显著意义的预测因子进行列线图的绘制(图7G),通过绘制分值的轴向垂线计算总分值,可以预测患者的1、 3、 5 年的生存率。进行GSEA富集分析评估预后模型相关的通路和功能。图8A列出了前7 个最有显著意义的通路,蛋白酶体、核糖体、嘌呤代谢、RNA聚合酶嘧啶代谢、戊糖磷酸途径和谷胱甘肽代谢途径富集在高风险组中,T细胞、B细胞受体信号通路、乙醚脂质代谢、醛固酮调节钠重吸收、原发性免疫缺陷、FCεR I信号通路以及JAK/STAT信号通路富集在低风险组。最后,使用HPA数据库评估基于免疫组化染色的预后基因表达水平(图8B)。结果显示大多数预后基因在正常组织中的表达量高于肿瘤组织,与TCGA的表达水平一致。
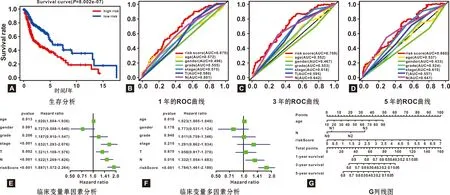
图7 预后模型的验证与列线图的构建
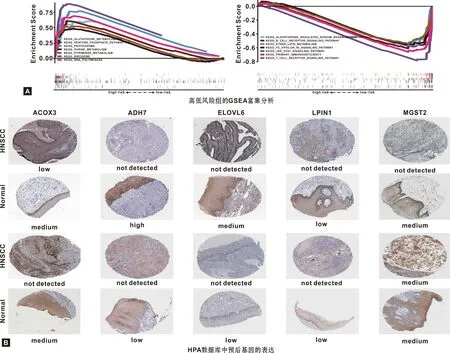
图8 GSEA富集分析及HPA数据库验证
3 讨 论
目前,高通量技术已广泛应用于HNSCC基因组、转录组和蛋白组水平的研究,但如今仍然缺少较为理想且稳定的生物标志物。因此,通过从公共数据库挖掘并整合高通量数据进而建立有效的预后标志物是有必要的。本研究通过利用R包WGCNA和limma分析GSE23036和E-MTAB-8588的芯片数据并得到60 个交集基因,在TCGA队列中进行单因素分析筛选出11 个基因与HNSCC生存相关。随后利用LASSO回归分析建立11 个基因的预后模型,单因素及多因素Cox分析证明风险评分是独立的预后因子,最后建立了一个列线图以辅助临床实践。
本研究中的基因预后模型包括11 个基因:EXPH5、ELOVL6、ACOX3、NAGK、LPIN1、TJP3、NBEAL2、SASH1、MGST2、TSPAN6和ADH7。EXPH5是一种Rab27b的效应蛋白,可以通过调控角质形成细胞调节表皮内稳态[12],在结直肠癌中发现其与COL1A2融合[13]。ELOVL6是超长链脂肪酸延伸酶,主要参与饱和与不饱和脂肪酸的延伸;其高表达与肝细胞癌的良好预后有关[14]。ACOX3是一种酰基辅酶A氧化酶,参与支链脂肪酸的代谢,被认为是宫颈癌的潜在生物标志物[15]。LIPIN1调节脂质和能量代谢,其过表达可促进乳腺癌进展[16]。NBEAL2是一种含有BEACH结构域的蛋白质家族成员,Wu等[17]曾研究报道NBEAL2与HNSCC的总体生存相关。TJP3是MAGUK蛋白超家族成员[18],被认为可促进卵巢癌细胞的侵袭和迁移[19]。SASH1是一种肿瘤抑制因子,抑制肿瘤细胞增殖、迁移及上皮间质转化[20];另一方面受MiR-9的负调控,促进HNSCC的肿瘤侵袭[21]。MGST2是谷胱甘肽S-转移酶(GST)基因家族成员[22],在UVB照射下发生甲基化并促进皮肤癌的进展[23]。乙醇脱氢酶(ADH)家族成员ADH7与非小细胞肺癌预后显著相关[24]。因此,本研究预后模型中的基因与多种癌症有关,以期为HNSCC的分子基础提供新的认识。
GSEA富集分析显示许多生物途径影响着肿瘤的发展。抑制蛋白酶体可抑制HNSCC细胞的生长[25]。循环外泌体中的嘌呤代谢水平影响着HNSCC的进展[26]。由ASCT2和GLUD组成的谷氨酰胺代谢相关基因可作为HNSCC靶向治疗的生物标志物[27]。抑制JAK/STAT通路抑制癌细胞增殖,可作为HNSCC有效的治疗手段[28]。因此,可进一步评估本研究中GSEA富集的通路在HNSCC中的作用。
综上所述,本研究中的风险模型可作为HNSCC的生物标志物,建立的列线图可指导临床医生的诊断及治疗。然而,本研究存在一定的局限性,需要临床试验评估风险模型的预后价值。
