星点设计效应面法优化葛根素改性壳聚糖纳米粒的处方与工艺研究
2023-11-30李思慧李娜关志宇徐鹏飞姜晟范瑾慧钟凌云朱卫丰江西中医药大学药学院南昌330004
★ 李思慧 李娜 关志宇 徐鹏飞 姜晟 范瑾慧 钟凌云 朱卫丰(江西中医药大学药学院 南昌 330004)
葛根素(Pur)药理活性广泛,具有抗炎[1]、抗过敏[2-3]和保护心血管系统[4]等作用,用于辅助治疗冠心病[5-8]、心绞痛[9-11]、缺血性脑卒[12-14]等疾病。人体内源性物质唾液酸能够参与细胞表面多种生理功能,在调节人体生理、生化功能方面起到非常重要的作用。通过唾液酸与血管内皮细胞表面特异性表达的E-selectin 受体结合,血管损伤时,特定炎症因子的表达,为血管靶向纳米粒的主动靶向提供了特异性与可行性[15-20]。本实验在前期对壳聚糖进行唾液酸修饰制备的改性壳聚糖(SA-CS)基础上,采用SA-CS 与三聚磷酸钠(TPP)制备葛根素改性壳聚糖纳米粒(SA-CS-Pur-NPs),并采用冷冻干燥技术制备葛根素冻干粉。通过单因素考察、星点设计效应面法优化制备SA-CSPur-NPs,选取常用的冻干保护剂对SA-CS-Pur-NPs 的冻干保护进行考察,以期筛选最优处方,提高葛根素纳米粒稳定性,为葛根素纳米粒的制备提供一定的研究基础,为临床应用及产业化生产奠定基础。
1 仪器与试剂
万分之一分析天平(塞多利斯科学仪器有限公司);超纯水仪(南京易普易达科技发展有限公司,EPED-ESL-10TH);超声仪(昆山市超声仪器有限公司,KQ5200DA);自动双重纯水整流器(上海亚荣生化仪器厂,SZ-93);马尔文纳米粒度仪(英国马尔文公司,Nano-S);高效液相色谱仪(美国安捷伦科技有限公司,Agilent A1260);磁力搅拌器(西安远舰仪器设备有限公司,ZNCLBS140×140);pH 校准计(上海仪电科学仪器股份有限公司,PHS-25 )。
葛根素对照品(中国食品药品检定研究院,批号:110752-201816,质量分数>95.5%);葛根素(成都普菲德生物技术有限公司,批号:18082401,质量分数>98%);CS-SA(实验室自制);PEG 2000(上海麦克林生化科技有限公司);TPP(上海易恩化学技术有限公司);色谱甲醇、色谱乙腈、色谱甲酸均为美国Tedia 公司。
2 方法与结果
2.1 SA-CS-Pur-NPs 制备方法
以SA-CS 和TPP 为载体,采用离子交联法制备SA-CS-Pur-NPs。首先称取葛根素10 mg,精密称定,超声溶于5%的乙醇水溶液,为备用溶液A;称取SA-CS 15 mg 溶于适量的纯水,得到2.5 mg/mL SA-CS 溶液,用稀盐酸与氢氧化钠溶液调节SA-CS溶液的pH 至6,为备用溶液B;配制5% PEG2000溶液为备用溶液C,0.5 mg/mL TPP 溶液为备用溶液D。然后精密量取A 溶液6 mL 于25 mL 烧杯,放置磁力搅拌器上,用注射器将5 mL 的B 溶液、1 mL的C 溶液依次缓慢滴加在A 溶液中,滴加完成后800 r/min 搅拌30 min,搅拌完成后将D 溶液滴加到上述溶液中,滴加完成后继续以800 r/min 搅拌30 min,得到供试品SA-CS-Pur-NPs 并进行测定。
2.2 葛根素的含量测定
2.2.1 色谱条件 色谱柱Agilent TC-C18(5 µm,4.6 mm×250 mm),流动相甲醇∶水=30∶70,流速为1 mL/min,检测波长为250 nm,柱温为30 ℃。
2.2.2 方法学考察 取5 mg 葛根素对照品配置成1 mg/mL 的储备溶液,分别取0.1,0.2,0.4,0.8,1.6,3.2 mL,分别置于 10 mL 量瓶中,加色谱甲醇定容至刻度线,按照“2.2.1”色谱条件进样,得到标准曲线Y=51.923X+231.11,R2=0.999 9,表明葛根素在10~320 µg/mL 线性关系良好。
取低、中、高(15、30、120 µg/mL)3 个质量浓度的葛根素对照品溶液,在“2.2.1”色谱条件下进样测定,每个浓度测定6 次,连续测定3 批。将样品的结果进行方差分析,根据样品求得本法的精密度,结果表明葛根素的低、中、高日内RSD值分别为1.01%、0.87%、0.69%,低、中、高日间RSD值分别为1.17%、1.81%、1.25%,说明该方法的精密度良好。精密量取1 mL 供试品溶液9 份,分为3 组,分别加入葛根素对照品溶液,得到加标质量浓度分别为低、中、高(15、30、120 µg/mL)的溶液,计算回收率分别为101.59%、99.44%、98.84%,RSD值分别为2.76%、1.00%、2.90%,表明回收率良好。取30 µg/mL 供试品溶液,分别在 0、2、4、6、8、12、18、24 h 进样分析,计算RSD值为2.68%。上述结果表明该方法可以用于葛根素纳米粒的含量测定。
2.2.3 包封率和载药量的测定 为了分离游离的葛根素,本实验采用超滤离心法测定纳米粒包封率(EE)和载药量(DL)。超滤离心法是将SA-CSPur-NPs 混悬液放入Nanosep® 10 KDa 超滤离心管上端,在12 000 r/min 下离心30 min,取续滤液按“2.2.1”下的色谱条件进行测定,计算EE、DL,计算公式如下:
C为混悬液中总的Pur 的质量浓度,C1为混悬液中未包封的药物的质量浓度,W1为包封的药物的质量,W2为所有辅料的质量。
2.2.4 粒径、PDI 和Zeta 电位的测定 精密量取SA-CS-Pur-NPs 1 mL,置于马尔文纳米粒度仪进行粒径、PDI 和Zeta 电位测定。
2.3 单因素考察
2.3.1 SA-CS 浓度的考察 称取葛根素10 mg,精密称定,TPP 浓度0.65 mg/mL,其他条件固定不变,考察CS 浓度为0.5、1.0、1.5、2.0、2.5、3.0 mg/mL 对SA-CS-Pur-NPs 粒径、PDI、电位、包封率、载药量的影响。结果显示,SA-CS 浓度在0.5 mg/mL 时,粒径>200 nm;1.0~3.0 mg/mL时,粒径在100~150 nm 之间,PDI 稳定且溶液澄清;1.0~2.5 mg/mL 时包封率>80%;随着SA-CS浓度的增大,载药量持续降低,综合各项评价指标,SA-CS浓度选择1.0~2.5 mg/mL进行处方优化,结果见表1。
表1 SA-CS浓度对粒径、PDI、电位、包封率、载药量的影响(,n=3)
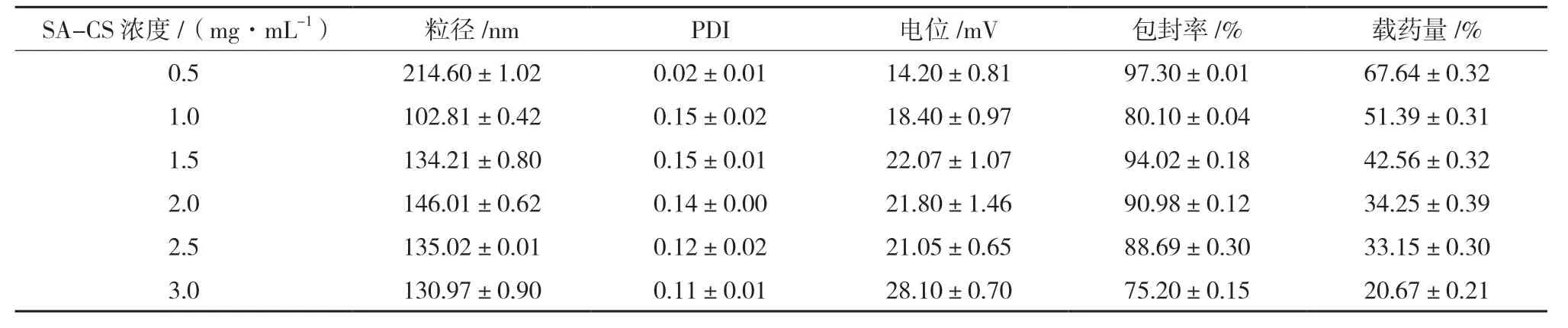
表1 SA-CS浓度对粒径、PDI、电位、包封率、载药量的影响(,n=3)
?
2.3.2 TPP 浓度的考察 确定了SA-CS 溶液的浓度之后,称取葛根素10 mg,精密称定,其他条件固定不变,考察TPP 浓度为0.50、0.65、1.00、1.50、2.00、2.50 mg/mL 对SA-CS-Pur-NPs 粒径、PDI、电位、包封率、载药量的影响。结果显示,TPP 浓度>1.00 mg/mL 时,混悬液有明显絮状物,且粒径快速增大,当TPP 浓度在0.50~1.00 mg/mL 时,粒径、PDI、包封率和载药量均为最佳,因此选择0.5~1 mg/mL 进行处方优化,结果见表2。
表2 TPP浓度对粒径、PDI、电位、包封率、载药量的影响(,n=3)

表2 TPP浓度对粒径、PDI、电位、包封率、载药量的影响(,n=3)
?
2.3.3 搅拌速度的考察 分别考察搅拌速度为200、400、600、800、1 000 r/min 时,搅拌转速对SA-CS-Pur-NPs 粒径、PDI、电位、包封率、载药量的影响。结果显示,搅拌速度低于400 r/min,反应不完全,致使混悬液的粒径过大,有明显沉淀,当搅拌转速大于800 r/min 时,包裹的药物会在高转速下与SA-CS 脱离,致使粒径增大,溶液不澄清,有较多絮状物悬浮。故选择转速为800 r/min 进行处方优化,结果见表3。
表3 搅拌速度对粒径、PDI、电位、包封率、载药量的影响(,n=3)
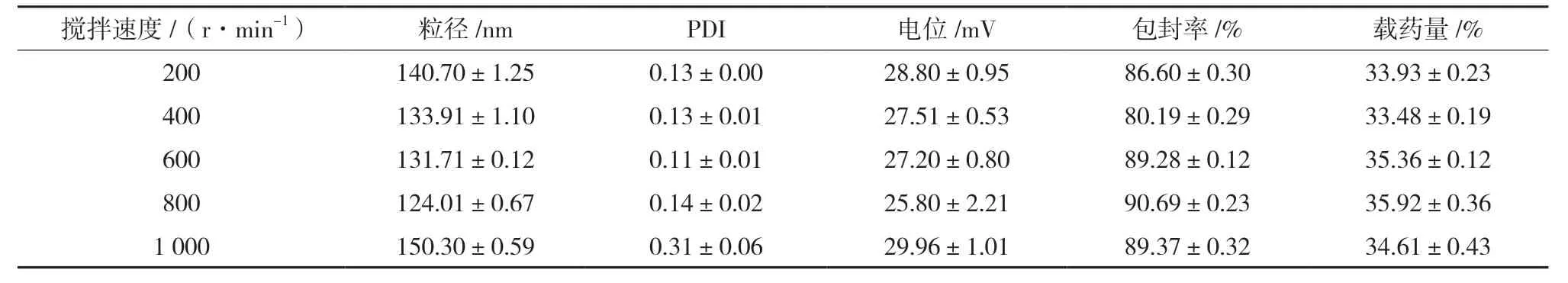
表3 搅拌速度对粒径、PDI、电位、包封率、载药量的影响(,n=3)
?
2.3.4 搅拌时间的考察 分别考察搅拌时间为15、30、60、90、120 min 时,搅拌时间对SA-CS-Pur-NPs 粒径、PDI、电位、包封率、载药量的影响。结果显示滴加TPP 后需搅拌一定的时间,混悬液有明显的乳光,但当搅拌时间过长,可能导致已形成的NPs 重新分散,使粒径变大,故将搅拌时间定为30 min,结果见表4。
表4 搅拌时间对粒径、PDI、电位、包封率、载药量的影响(,n=3)
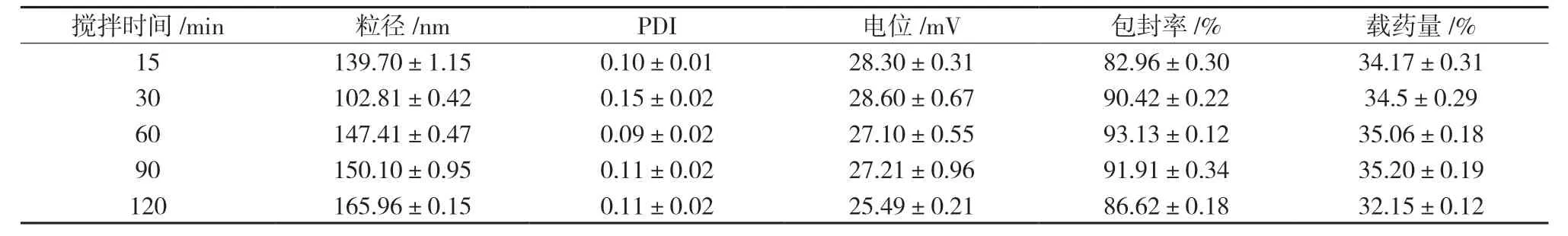
表4 搅拌时间对粒径、PDI、电位、包封率、载药量的影响(,n=3)
?
2.3.5 温度的考察 分别考察搅拌温度为20、25、30、35、40 ℃时,搅拌温度对SA-CS-Pur-NPs 粒径、PDI、电位、包封率、载药量的影响。结果显示,升高温度会使纳米粒粒径增大,包封率、载药量则降低。因为温度升高,在纳米尺寸的颗粒间相互作用力比较大,容易发生团聚,导致粒径增大。故将温度定为室温,结果见表5。
表5 温度对粒径、PDI、电位、包封率、载药量的影响(,n=3)
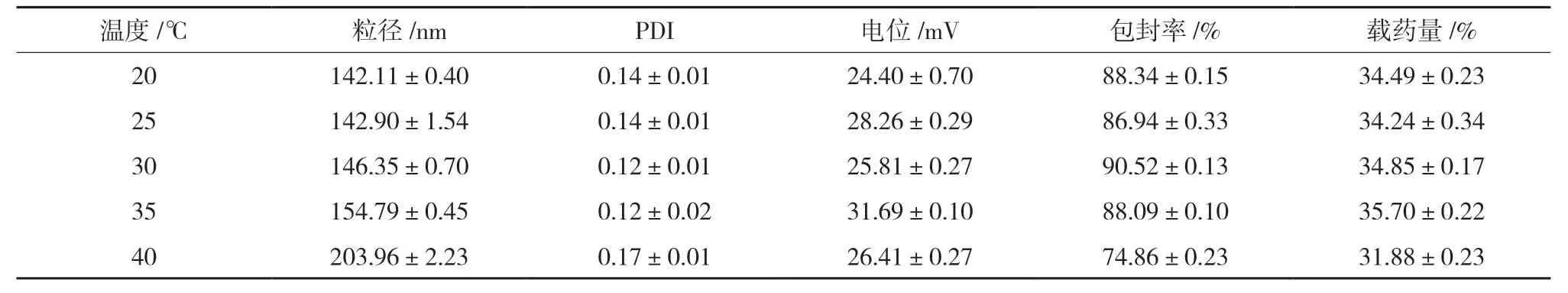
表5 温度对粒径、PDI、电位、包封率、载药量的影响(,n=3)
?
2.3.6 pH 的考察 确定了SA-CS 最佳浓度之后精密称取Pur 10 mg,TPP 浓度0.65 mg/mL,其他条件固定不变,用稀盐酸和饱和氢氧化钠溶液调节SA-CS 溶液的pH 值,分别考察SA-CS 溶液的pH为 4.0、4.5、5.0、5.5、6.0、6.5 对SA-CS-Pur-NPs 粒径、PDI、电位、包封率、载药量的影响。综合评价调节pH 对SA-CS-Pur-NPs 的粒径、电位、包封率和载药量的影响,且通过测定SA-CS 溶液的pH 在6.0~6.5 的范围内,为防止过酸或过碱对SA-CS 造成解离,影响纳米粒的形成,因此选择5.0~6.5 的pH 区间进行处方优化,结果见表6。
表6 pH对粒径、PDI、电位、包封率、载药量的影响(,n=3)
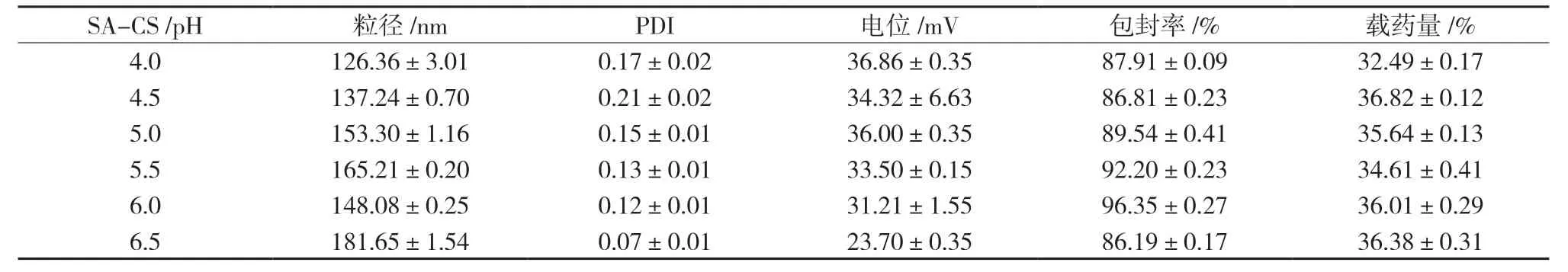
表6 pH对粒径、PDI、电位、包封率、载药量的影响(,n=3)
?
2.3.7 PEG 2000 加入量考察 精密称取Pur 10 mg,TPP 浓度0.65 mg/mL,其他条件固定不变,考察PEG 2000 加入量为5%、10%、15%、20%、25%时对SA-CS-Pur-NPs 粒径、PDI、电位、包封率、载药量的影响。结果显示,PEG 2000 的加入量在5%~25%的范围内,粒径、电位均为最佳,结果见表7。
表7 PEG 2000加入量对粒径、PDI、电位、包封率、载药量的影响(,n=3)
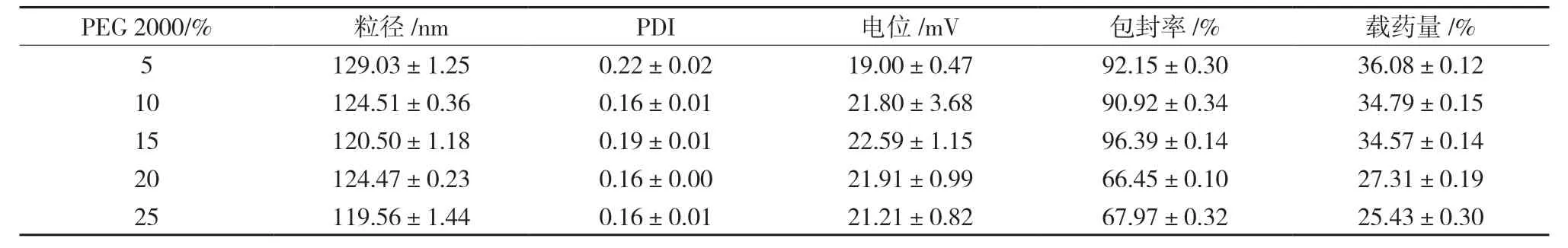
表7 PEG 2000加入量对粒径、PDI、电位、包封率、载药量的影响(,n=3)
?
2.4 中心复合设计效应面法优化纳米粒处方
依据单因素实验结果,采用中心效应面法对离子凝胶化法制备 SA-CS-Pur-NPs 进行处方优化,选取因素A 为SA-CS 浓度(mg/mL)、B 为TPP 浓度(mg/mL)、C 为SA-CS pH、D 为PEG 2000(%),试验设计见表8。
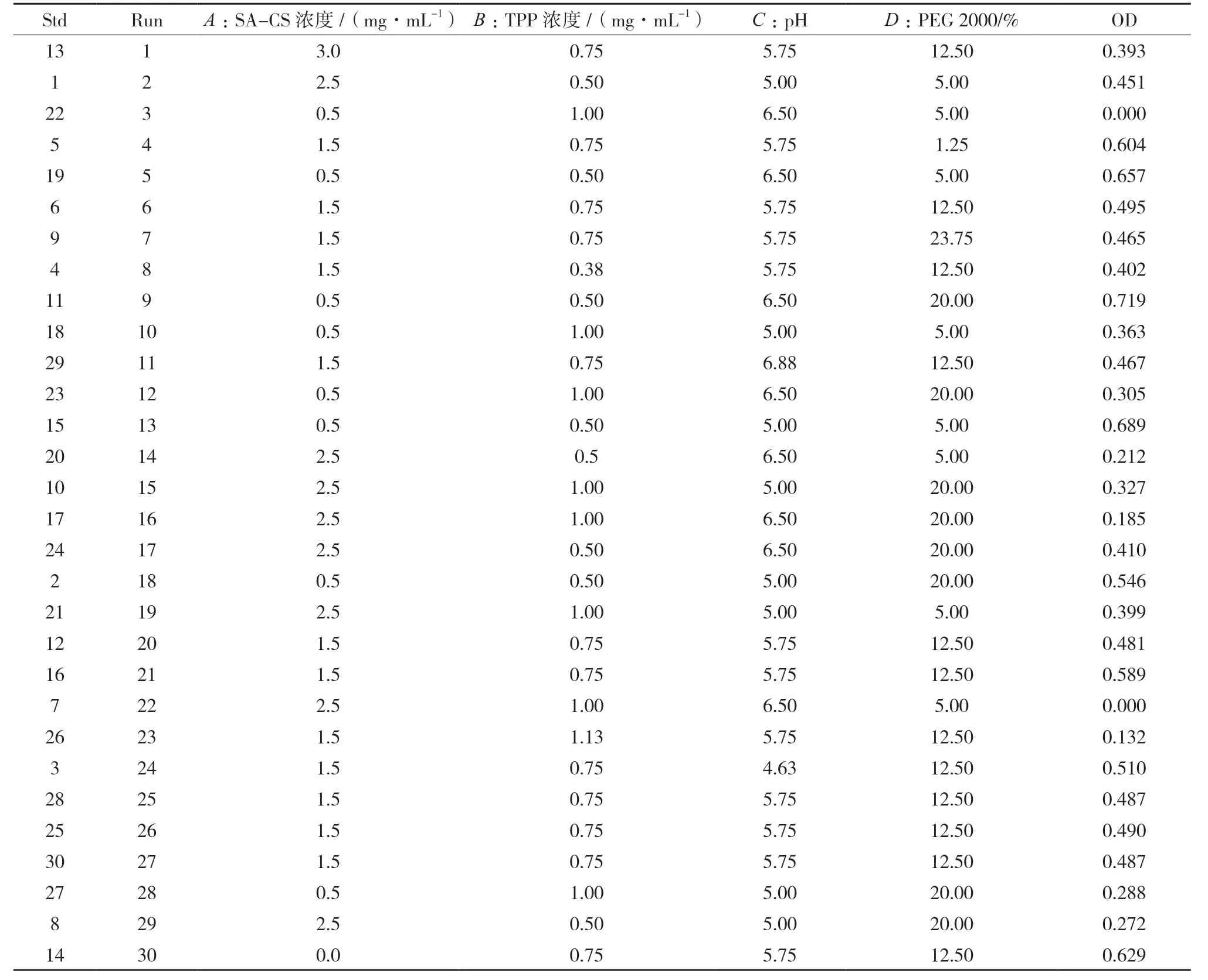
表8 SA-CS-Pur-NPs中心复合设计安排与结果(n=3)
根据 Design Expert 8.0.6 软件,将平均粒径、多分散系数(PDI)、包封率、载药量每个指标定为0~1 的归一值,各指标归一值求几何平均数,得总评归一值(OD),按公式 OD =(d1d2…dk)1/k进行计算,k 为指标数[21-22]。对于包封率、载药量取值越大越好,平均粒径、PDI 取值越小越好,分别采用Hassan 方法,然后分别转换求得归一值dmax和dmin公式如下[23]。星点设计效应面法试验结果见表8,所得OD 值各因素方差分析结果见表9。
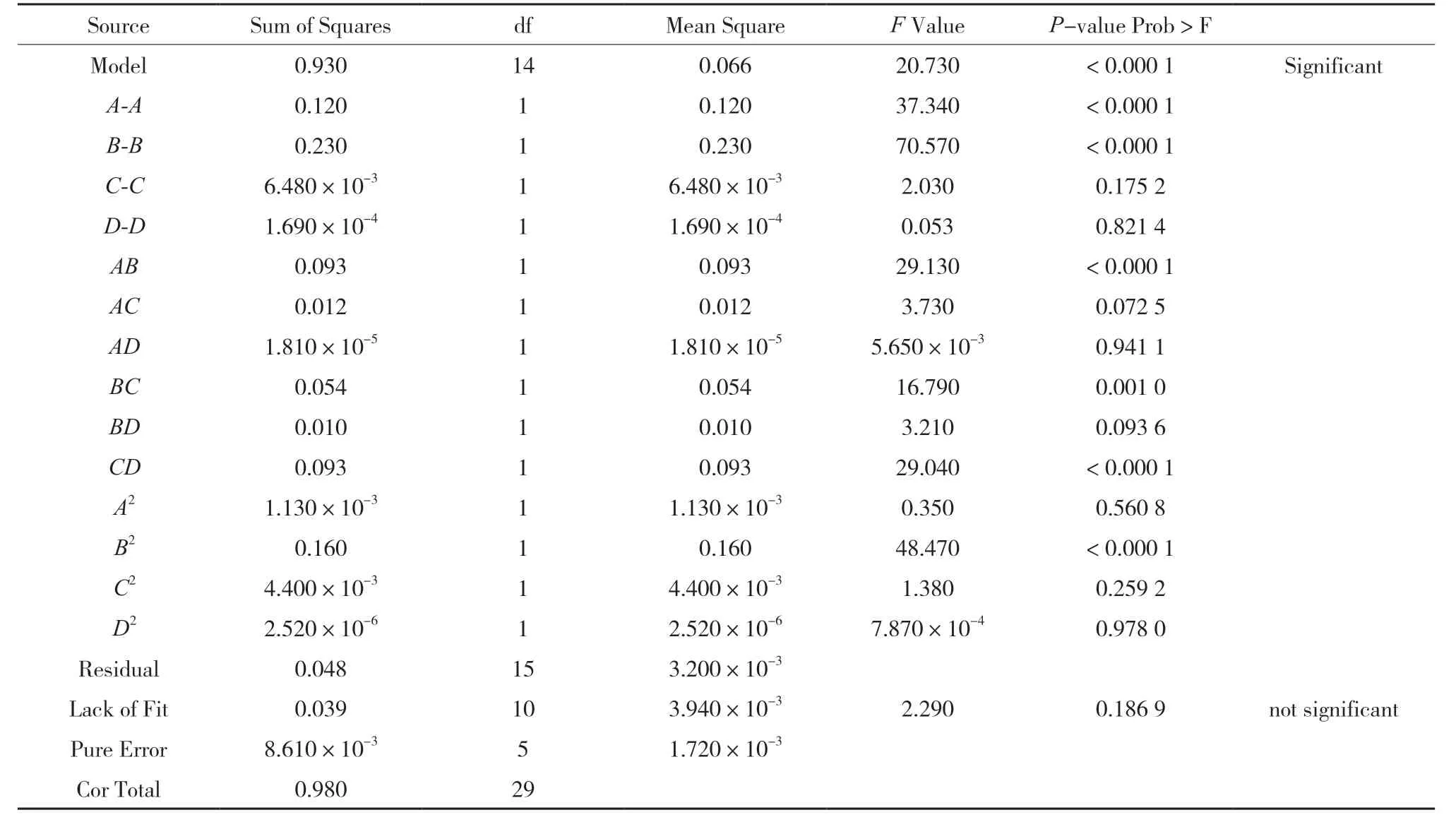
表9 各因素回归系数和方差分析
应用Design Expert 8.0.6 软件以A、B、C、D为自变量,OD 值为因变量,进行2 次多项式逐步回归拟合,得到方程R2=0.52-0.12A-0.11B-0.019C+5.134×10-3D+0.11AB-0.041AC-2.125×10-33AD-0.058BC+0.034BD+0.10CD-0.022A2-0.12B2-0.020C2+8.362×10-3D2,其等高线图与3D 曲线图见图1。中心效应面法得到最优处方:SA-CS 浓度为2.5 mg/mL,SA-CS 的pH 为6.05,TPP 浓度为0.50 mg/mL,PEG的浓度为5%。
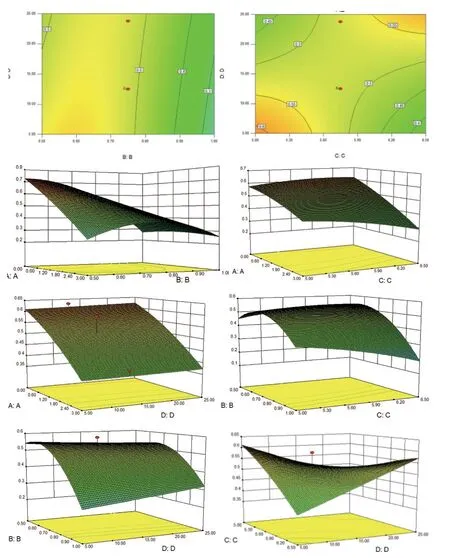
图1 效应值 OD 与 A、B、C、D 4 个因素的等高线图和三维曲面图
2.5 最优处方验证实验
根据优化后的处方和工艺,进行重复验证实验,为了方便实验操作,将最优处方和工艺定为SA-CS 浓度为2.5 mg/mL,SA-CS 的pH 为6.00,TPP 浓度为0.50 mg/mL,PEG 的浓度为5%。将效应面得到的最佳处方和工艺进行3 次平行实验,粒径、PDI、包封率、载药量4 个指标的预测值与实测值的RSD分别为0.21%、0.66%、0.12%、0.62%,表明相对于预测值而言,实测值与其并无显著性差异,即建立的回归方程预测性良好,结果见表10。

表10 3次平行实验后预测值与实测值之间的比较(n=3)
2.6 冻干技术考察
2.6.1 冻干保护剂考察 选取常用的冻干保护剂甘露醇、蔗糖、乳糖和葡萄糖对SA-CS-Pur-NPs 的冻干保护进行考察,取适量新制SA-CS-Pur-NPs混悬液至表面皿中,加入等体积等质量浓度的冻干保护剂溶液,将两者混匀,于-20 ℃低温冰箱中分别预冻24 h 后立即进行冷冻干燥。对照组则以纯水替代冻干保护剂溶液,其他条件一致。所得冻干粉加入原体积纯水震摇,考察冻干粉的复溶性。同时测定冻干粉复溶后混悬液的粒径、电位、PDI、包封率和载药量。
观察冻干样品的状态,测定各指标的变化情况,结果见表11。从外观看,甘露醇组的保护纳米粒质地松软,细腻,结构完整,用纯水复溶后无结块,其他组均存在肉眼可见的不溶颗粒。且综合复溶后各保护剂冻干后的纳米粒,其粒径、电位、包封率、载药量等各项指标来看,保护剂为单个的甘露醇时为最佳。
表11 不同保护剂样品复溶后各项指标(,n=3)
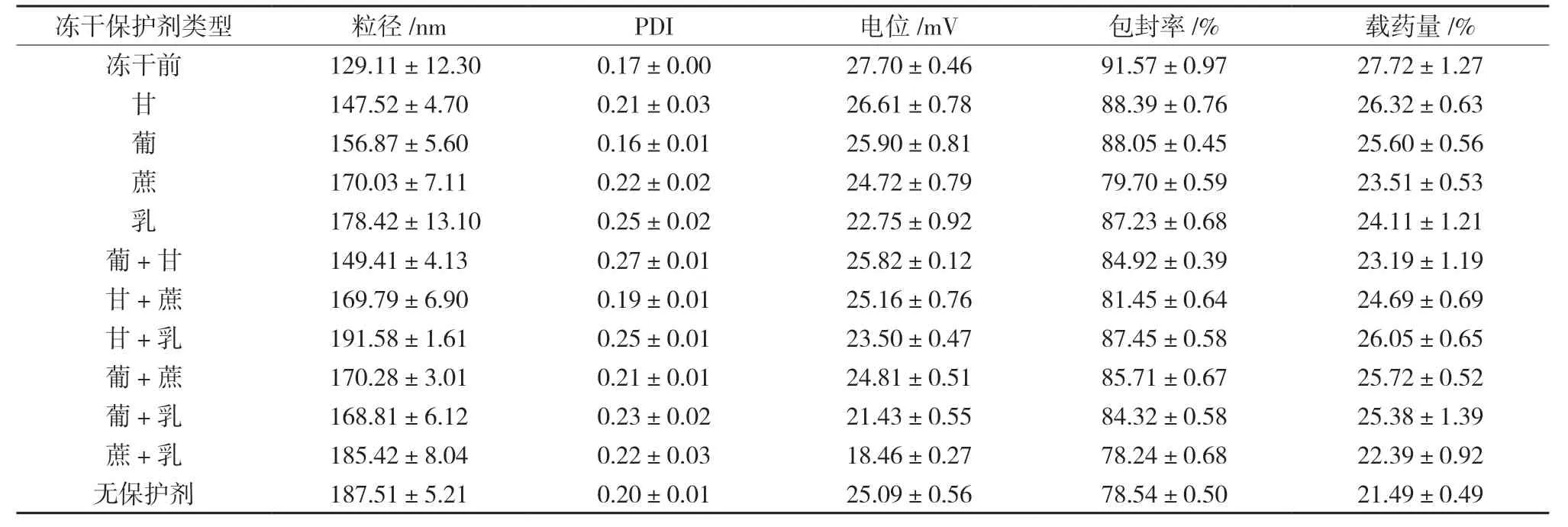
表11 不同保护剂样品复溶后各项指标(,n=3)
?
表12 不同浓度甘露醇复溶后各项指标(,n=3)
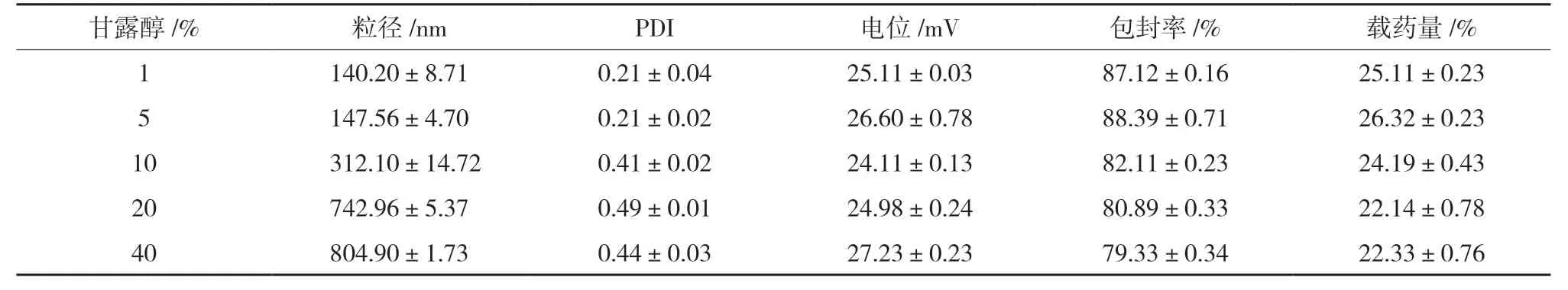
表12 不同浓度甘露醇复溶后各项指标(,n=3)
?
2.6.2 甘露醇用量考察 选取甘露醇做冻干保护剂,取适量新制SA-CS-Pur-NPs混悬液至表面皿中,加入等体积质量分数分别为1%,5%,10%,20%,40%的冻干保护剂溶液,结果显示1%、5%复溶后溶液是澄清的,10%以上的甘露醇均存在肉眼可见的不溶颗粒。而复溶后10%以上的甘露醇会导致纳米粒的粒径及PDI 值都增大,不符合要求。当甘露醇大于5%时外观平整无气泡冻干效果好,故选取5%的甘露醇进行冻干保护。
2.7 冻干工艺验证
根据优化后的处方工艺,进行3 次平行实验。冻干后复溶的粒径为(147.50±4.70)nm,PDI 为(0.21±0.03),电位为(26.60±0.78)mv,包封率为(88.39±0.76)%,载药量为(26.32±0.63)%。证明该工艺稳定可行。
3 讨论
本文采用离子交联法制备SA-CS-Pur-NPs,通过葛根素、SA-CS、TPP 与PEG 2000 结构中离子的相互交联作用形成纳米粒。制备中通过对纳米粒形成过程中的各项因素进行单因素考察筛选出显著影响纳米粒形成的因素,再通过星点设计效应面对该纳米粒的处方进行进一步的优化设计,确定了最佳处方与工艺,成功制备了粒径在130 nm 左右的SA-CS-Pur-NPs,结构稳定,易于制备。其中影响SA-CS-Pur-NPs 粒径最主要的因素为TPP 的浓度和SA-CS的pH,当TPP的浓度>1.00 mg/mL时,SA-CS-Pur-NPs 的粒径迅速增大,溶液浑浊,有絮状沉淀产生,整个纳米粒体系及其不稳定,TPP 溶液的浓度对纳米粒的形成影响非常大。而SA-CS的pH 的考察,除了考虑在纳米粒形成中的影响之外,还应考虑在SA-CS 的合成过程中,应避免过酸与过碱的情况以及SA-CS 的解离。优选出5%甘露醇为冻干保护剂制得的冻干粉用水分散后,其外观为淡蓝色半透明液体,粒径、电位、包封率、载药量等各项指标无明显改变,较稳定。本文优选出的葛根素改性壳聚糖纳米粒冻干粉工艺,外观饱满,复溶性好,稳定性得到显著提高。
