一株H1N1亚型猪流感病毒全基因组特征分析
2023-11-30刘可欣刘佳利张淑琴
张 傲,谭 斌,刘可欣,刘佳利,张淑琴
(中国农业科学院特产研究所,长春 130112)
猪流感病毒(swine influenza virus,SIV)为正黏病毒科、A型流感病毒属成员,可引起猪的急性、热性、高度传染性呼吸系统疾病。目前,世界范围内主要存在的SIV包括H1N1、H1N2和H3N2亚型[1]。猪流感病毒变异速度快,由于猪呼吸道黏膜上皮细胞表面同时存在禽流感病毒和人流感病毒的唾液酸受体,使得不同来源的流感病毒可在猪体内进行基因重排,产生具有新的感染性、致病性及传播性的基因重排毒株,不仅给全球养殖业带来了巨大的经济损失,并且对公共卫生造成了严重危害[2-5]。因此,加强对SIV的流行病学调查,及时掌握SIV的生物学特性及流行情况,不仅对SIV的预防和控制具有重要作用,更具有深远的公共卫生学意义。本研究在吉林地区某猪场分离得到一株H1N1亚型SIV,通过对该分离株进行全基因组特征分析,了解SIV在吉林地区猪场中的流行情况,为我国猪流感防控提供参考依据。
1 材料与方法
1.1 主要试剂材料
犬肾细胞(MDCK)由中国农业科学院特产研究所保存;SPF鸡胚购自北京梅里亚维通实验动物技术有限公司;Trizol购自莫纳(长春)生物科技有限公司;DNA Marker、Trans1-T1感受态细胞均购自北京全式金生物技术有限公司;胶回收试剂盒购自OMEGA公司;PCR试剂、克隆载体pMD18-T均购自TaKaRa公司。
1.2 病毒的分离鉴定
将采集的鼻拭子加入1 mL双抗PBS溶液(2 000 U·mL-1)稀释震荡,在4 ℃条件下8 000 r·min-1离心10 min,取上清以0.22 μm滤膜滤过除菌,接种于9~10日龄SPF鸡胚尿囊腔中,37 ℃培养72 h后收集鸡胚尿囊液。按照上述方法将尿囊液再次接种鸡胚盲传2代,收集第3代尿囊液进行红细胞凝集试验,测定其对1%鸡红细胞的凝集活性并通过RT-PCR鉴定SIV。对鉴定结果呈阳性的尿囊液参照GB/T 27521—2011猪流感病毒核酸RT-PCR检测方法对分离病毒进行PCR分型鉴定,并按照Reed-Muench法测定病毒的TCID50。
1.3 病毒全基因组扩增及特征分析
根据GenBank查询到的H1N1亚型SIV基因序列信息,通过比对参考毒株的全基因组序列,设计扩增引物。应用Trizol提取分离株的RNA,通过RT- PCR扩增全基因序列并连接至pMD18-T载体后送至生工生物工程(上海)有限公司进行测序。测序结果采用BLAST和MegAlign进行序列比对及同源性分析,运用MEGAX绘制系统进化树,NetNGlyc-1.0分析预测潜在糖基化位点。
1.4 吉林省H1N1 SIV的选择压力分析
根据NCBI查询到的全部吉林省H1N1亚型SIV基因序列信息,结合分离株的基因序列,采用Datamonkey中的SLAC(Singlelikelihood ancestor counting)及MEME(mixed effects model of evolution)方法计算每个密码子的非同义替换(dN)与同义替换(dS),分析吉林省H1N1 SIVNA和HA基因的选择压力、阴性选择位点和阳性选择位点。毒株检索时间区间为2008—2022年,数据库采集数据时间为2023年3月9日。P<0.1时,SLAC和MEME的计算结果具有显著性。
2 结 果
2.1 病毒的分离鉴定
收获鸡胚尿囊液进行红细胞凝集试验,结果显示,有12份鸡胚尿囊液具有血凝性,其中最高血凝价为1∶128。取最高血凝价分离毒株进行后续试验,将该毒株命名为A/swine/Jilin/25/2008(H1N1)。利用亚型鉴定引物对分离病毒进行PCR分型鉴定,结果显示分离株为H1N1亚型SIV。TCID50测定结果显示分离株在MDCK 细胞上的TCID50为10-4.88·mL-1。
2.2 病毒全基因组序列特征分析
2.2.1 病毒全基因组同源性分析 分离株全基因组的8个基因节段(GenBank登录号为OQ740141~OQ740148)与GenBank登录的基因序列比对结果显示,HA基因与A/turkey/IA/21089-3/1992(H1N1)核苷酸相似性为98.94%;NA基因与A/swine/Iowa/24297/1991(H1N1)核苷酸相似性为99.29%。内部基因PB2、PA、NP、M、NS基因与2008年在中国出现的A/swine/Hubei/02/2008(H1N1)病毒具有高度相似性,核苷酸序列相似性为98.58%~99.64%;PB1基因与A/swine/Iowa/24297/1991(H1N1)核苷酸相似性最高,为99.49%。
2.2.2 HA 序列分析 HA蛋白由SIV第4节段的RNA编码,其开放阅读框(open reading frame,ORF)全长为1 701 bp,共编码566个氨基酸,其中HA1由326个氨基酸组成,HA2由223个氨基酸组成。利用SignalP 4.0信号肽预测软件对分离株HA蛋白进行信号肽预测,结果显示HA信号肽序列位于1~17位氨基酸。经Lasergene分析,HA基因的裂解位点位于339~347位氨基酸处,序列为339PSIQSR↓GLF347,该位点未发生氨基酸突变或插入,符合低致病性SIV的分子特征。利用NetNGlyc-1.0预测HA潜在糖基化位点,结果显示HA蛋白具有6个潜在的糖基化位点,分别为28NSTD、40NVTV、104NGTC、304NTSL、498NGTY和557NGSL。
2.2.3 NA 序列分析 NA蛋白由SIV第6节段的RNA编码,其ORF全长为1 410 bp,共编码470个氨基酸。其潜在糖基化位点有6个,分别为44NHSE、63NOTY、68NISN、88NSSL、146NGTV和235NGSC。通过对NA氨基酸序列分析发现,分离株出现H275K抗神经氨酸酶抑制剂(neuraminidase inhibitor,NAI)耐药性相关氨基酸位点突变,表明该病毒可能对奥司他韦(oseltamivir)等NAI具有抗药性[6-7]。
2.2.4 病毒HA基因和NA基因遗传进化分析 将分离株的8个基因节段核苷酸序列与GenBank中的经典型H1N1、欧亚类禽型H1N1、类人型H1N1和2009H1N1分支代表毒株进行比对分析(图1)。结果显示,分离株的全基因组均属于经典型H1N1进化分支,未出现流感病毒不同基因型之间的基因重排。
2.2.5 分离株功能性氨基酸位点突变分析 对分离株进行功能性氨基酸位点突变分析,毒株出现SIV致病性、毒力及复制能力增强的相关位点突变,结果见表1。
2.3 吉林省H1N1 SIV的选择压力分析
通过SLAC和MEME方法对NCBI检索到的吉林省现有H1N1 SIV毒株及本研究分离得到的H1N1 SIV进行选择压力分析,HA筛选出4个正向选择位点及20个负向选择位点,NA筛选出6个正向选择位点及11个负向选择位点,二者的选择压力分别为0.123和0.138,结果见表2。SIVHA和NA基因在吉林省的自主变异进化明显,NA的突变率高于HA。
3 讨 论
SIV可引起猪的高度传染性呼吸系统疾病,该病毒的传播给我国畜牧业造成了巨大的经济损失,并对人类构成潜在的大流行威胁。自1918年以来,经典型H1N1 SIV在猪群中传播并以重组的形式在人类中出现,导致了2009年H1N1流感大流行[8]。本研究对分离出的经典型H1N1 SIV全基因组进行了系统分析,并对吉林省H1N1 SIV的选择压力进行了分析评估。
之前的研究报道中指出,直至1998年,经典型H1N1 SIV作为主要的流行毒株在北美猪群中持续传播。在1918—1919年大流行期间,经典型H1N1 SIV可能由北美传播至中国沿海城市的猪群中,该病毒于1991年首次从中国大陆分离出来,流感监测分析显示,自1996年以来,我国猪群中经常分离出经典型H1N1 SIV[9-10]。本研究于2008年在吉林地区分离得到一株经典型H1N1 SIV,并命名为A/swine/Jilin/25/2008(H1N1)。同源性和系统发育分析表明,该病毒与此前在北美和中国分离的经典型H1N1病毒有着密切的亲缘关系,这表明它们可能属于同一进化分支,与之前流感病毒研究与监测分析结果相一致。
先前的研究表明,H1N1亚型SIV对NAI具有高耐药性的突变为H275Y,该位点突变使NA活性位点结构发生改变,并降低了NAI与NA的结合[7-8]。本研究中分离株出现H1N1亚型SIV NAI高耐药性氨基酸位点突变,表明该病毒可能对奥司他韦等NAI具有抗药性。目前,仅有少数毒株具有神经氨酸酶抑制性突变,故该毒株的序列分析对后续抗流感药物研究具有参考意义。
由于SIV聚合酶复合物缺乏校对活性,HA和NA基因易发生点突变,常以dN/ dS评估氨基酸位点所受的自然选择压力[11]。自发突变在负向选择压力下会被清除,而在正向选择压力下则可以积累并稳定遗传,导致蛋白功能的变化。本研究筛选出
HA基因及NA基因较多的负向选择位点表明SIVHA和NA基因在吉林省的自主变异进化明显,已超越免疫选择压力的作用。在我国猪的寿命约为6个月,且猪场几乎不使用流感疫苗,流感病毒被引入猪体内后,病毒所受的免疫选择压力较小。因此,这些基因的自主变异进化会超越免疫选择压力的作用,导致其进化速度比人类和禽类流感病毒慢。
表1 分离株功能性氨基酸位点突变分析Table 1 Analysis of functional amino acid mutations of the SIV isolates
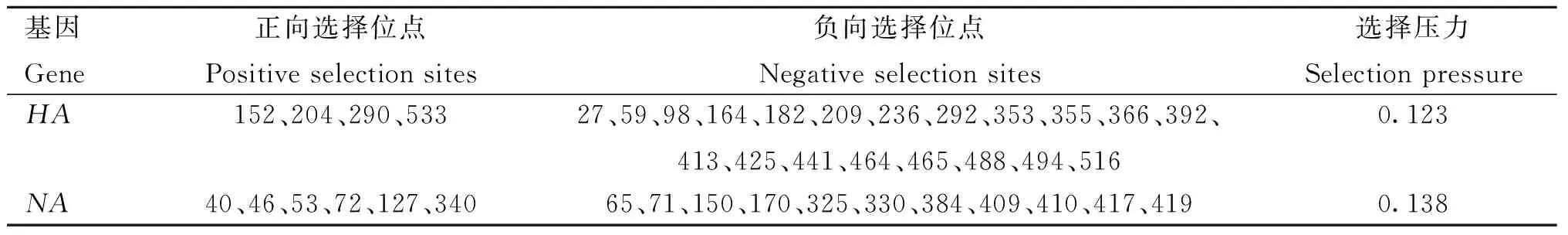
表2 吉林省H1N1 SIVs的选择压力分析Table 2 Analysis of the selection pressure of Jilin H1N1 SIVs
4 结 论
本研究成功分离出一株SIV,通过对分离株全基因组进行系统分析确认该毒株为经典型H1N1 SIV,并命名为A/swine/Jilin/25/2008(H1N1)。本研究获得的H1N1亚型SIV分离株全基因组序列分析结果对SIV流行病学调查及后续疫苗研发具有一定意义。
