Neurologic orphan diseases: Emerging innovations and role for genetic treatments
2023-11-29IvelinaKioutchoukovaDevonFosterRajviThakkarMarooForemanBrandonBurgessRebeccaTomsEduardoMolinaValeroBrandonLuckeWold
Ivelina P Kioutchoukova, Devon T Foster, Rajvi N Thakkar, Maroo A Foreman, Brandon J Burgess, Rebecca M Toms, Eduardo E Molina Valero, Brandon Lucke-Wold
Abstract Orphan diseases are rare diseases that affect less than 200000 individuals within the United States.Most orphan diseases are of neurologic and genetic origin.With the current advances in technology, more funding has been devoted to developing therapeutic agents for patients with these conditions.In our review, we highlight emerging options for patients with neurologic orphan diseases,specifically including diseases resulting in muscular deterioration, epilepsy,seizures, neurodegenerative movement disorders, inhibited cognitive development, neuron deterioration, and tumors.After extensive literature review, gene therapy offers a promising route for the treatment of neurologic orphan diseases.The use of clustered regularly interspaced palindromic repeats/Cas9 has demonstrated positive results in experiments investigating its role in several diseases.Additionally, the use of adeno-associated viral vectors has shown improvement in survival, motor function, and developmental milestones, while also demonstrating reversal of sensory ataxia and cardiomyopathy in Friedreich ataxia patients.Antisense oligonucleotides have also been used in some neurologic orphan diseases with positive outcomes.Mammalian target of rapamycin inhibitors are currently being investigated and have reduced abnormal cell growth, proliferation, and angiogenesis.Emerging innovations and the role of genetic treatments open a new window of opportunity for the treatment of neurologic orphan diseases.
Key Words: Neurologic orphan diseases; Gene therapy; Clustered regularly interspaced palindromic repeats/Cas9; Antisense oligonucleotides; Adeno-associated virus; mTOR inhibitors
lNTRODUCTlON
Orphan diseases are rare diseases that affect less than 200000 individuals within the United States[1].Despite being rare,these diseases affect over 300 million individuals globally[2].Of the 7000 diseases listed on the National Institute of Health's Office of Rare Diseases site, most are neurological and of genetic origin[3].90% of orphan diseases have serious neurological effects[2].Neurologic orphan diseases are fatal, drastically decrease quality of life, and are defined by long periods of disability[4].However, diagnosis and treatment of rare central nervous system (CNS) disorders pose a challenge[2].Lack of access to diagnostic genomic sequencing, screening tests, and specialists contributes to the difficulty of diagnosing and managing neurologic orphan diseases[2].Most neurologic orphan diseases don’t have treatments that prevent disease progression[4].Additionally, clinical trials investigating neurologic orphan disease therapeutics have the lowest success rate[5].Global Genes, a rare disease advocacy organization, states that since 2021, a total of $22.9 billion has been invested in research on neurologic orphan diseases, which offers a promising future for patients with these conditions[6].
One study found that the use of whole genome sequencing within clinical practice increases the diagnosis of neurologic orphan diseases[7].Genetic conditions such as Huntington's disease and Friedreich's ataxia are caused by repeat expansions which can effectively be detected by whole genome sequencing due to its high sensitivity and specificity for repeat expansions[7].
The Patient Identification and Engagement for Rare CNS Disorders initiative is designed to investigate and improve barriers to diagnosis and clinical research trials[2].This initiative plans to address the underrepresentation of certain groups from clinical trials and to improve access for those individuals in the participation of trials investigating new gene therapy approaches[2].The future for the diagnosis and management of neurologic orphan diseases looks promising and hopes to improve the efficacy of therapeutic agents by gene targeting methods.Some forms of gene therapy include the use of clustered regularly interspaced palindromic repeats (CRISPR)/Cas9, antisense oligonucleotides (ASO), adenoassociated viruses (AAV), and mammalian target of rapamycin (mTOR) inhibitors.
CRISPR/Cas9 is a form of gene-editing where a guide RNA binds to a target sequence of genomic DNA, followed by the endonuclease, Cas9, binding to the guide RNA.Cas9 then creates a double strand break in the genomic DNA which is then repaired.This process allows for the inclusion or exclusion of desired genes to create a desired mutation[8].
ASO are small molecules that can modify gene expression preventing or altering protein production.If a certain protein is undesired, an ASO can be designed to cause the protein to be terminated or partially expressed and modify it so that it is not harmful[9].
AAV are used as a vector for gene therapy by using a non-enveloped virus engineered to deliver deoxyribonucleic acid(DNA) to targeted cells[10].This gene therapy has shown preclinical and clinical access in gene replacement, gene silencing, and gene editing[11].
mTOR is a kinase closely correlated with the occurrence of neurodegenerative diseases and tumors in humans.The goal of mTOR inhibition therapies is to block the mTOR signaling pathway that may be contributing to abnormal signal transduction to block the occurrence and development of disease[12].
MUSCULAR DETERlORATlON
Spinal muscular atrophy
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disease caused by deletions or mutations in the survival motor neuron (SMN1) gene[13].Specifically, diagnostic testing commonly demonstrates the absence of SMN1 exon 7 on chromosome 5[14,15].SMA is characterized by progressive muscle weakness and atrophy resulting from progressive degeneration and irreversible loss of anterior horn cells in the spinal cord[16].The severity of SMA can range from more mild cases where the onset occurs in adulthood and progresses at a slow rate to more severe cases where the onset can occur in the first months of life and result in respiratory failure[17].
In recent years, new treatment options like gene therapy involving splicing modulation of SMN2 and SMN1 genes and the development of the first approved drugs for SMA treatment have shown promise in treating SMA.When therapy is initiated early, it can significantly alter the natural course of the disease.Current evidence in these treatments is limited to a small scope of patients and more research is needed for conclusive results[17].
Nusinersen was the first drug that received approval for the treatment of SMA.Nusinersen enhances the inclusion of exon 7 in mRNA transcripts of SMN2 by suppressing the binding of certain splicing factors which results in an increase in functional SMN2-mRNA with included exon 7[18-20].Various studies in infants and young children have displayed improvements in prolonged time of death and improved motor functions[21-23].Furthering the potential of SMN2 gene alteration, Risdiplam, and Branaplam are oral medications that have been shown to cross the blood-brain barrier and increase the number of full-length SMN proteins[24].
Gene therapy has also shown promise with SMN1-gene replacement.Studies in mice have shown prolonged survival following successful vector delivery of intact SMN1-gene across the blood-brain barrier[25,26].Zolgensma is a gene therapy medicine that is administered as an intravenous infusion and uses adeno-associated virus vectors to deliver a functional copy of SMN to motor neuron cells[27].Clinical trials in children treated with Zolgensma have shown improved survival, motor function, and developmental milestones following treatment[28,29].A summary of emerging treatment options for SMA can be found in Figure 1 below.
Duchenne muscular dystrophy
Duchenne muscular dystrophy (DMD) is the most common hereditary neuromuscular disease and is one of the most severe forms of inherited muscular dystrophy.Symptoms of DMD include severe and progressive muscle wasting,muscle weakness, and difficulty with movement[30].Late stages of DMD often require the need for assisted ventilation and the use of a wheelchair to perform daily activities and lead to premature death in the mid-twenties due to respiratory muscle weakness or cardiomyopathy[31].
DMD is an X-linked inherited disorder that predominantly affects males.The onset of DMD occurs due to mutations in the dystrophin gene on chromosome Xp21.This results in a ceased production of dystrophin in cardiac and skeletal muscle.The absence of dystrophin results in a loss of myofibril membrane integrity through cycles of necrosis and regeneration.Fibrous connective tissue and fat then replace muscle over time, resulting in the progression of expressed clinical symptoms[30,32].
In recent years, there has been progress in the development of diagnosis and therapeutics for DMD, but the current treatments given do not cure DMD[33].Daily prednisone treatment is commonly used to increase muscle strength and function, improve pulmonary function, and significantly slow the progression of weakness[34,35].While this treatment does not cure the disease, it does improve the overall quality of life for patients.
The most advanced therapy is antisense oligonucleotides -mediated exon skipping which has shown promise in clinical application.For this technique, the administration of 20-30 bp long antisense oligonucleotide hybridizes to splice motifs necessary for pre-mRNA processing and mask RNA splicing signals.This leads to the exclusion of the intron and adjacent exon, creating an in-frame mRNA without the targeted exon.This mRNA can then be translated into a truncated and partially functional dystrophin protein[33,36].
CRISPR/Cas technology has been used therapeutically to treat DMD by upregulating a dystrophin homolog, utrophin,to compensate for the lack of dystrophin protein which has been successfully demonstrated in patient cells.In these patients, full-length dystrophin was restored in patient cells carrying duplication mutations[37].Manipulations resulting from CRISPR-Cas9 were shown to restore the expression of truncated but partially functional dystrophin, improve skeletal and cardiac muscle function, and increase the survival of mdx mice significantly[38].
Emerging vector-mediated gene therapy in mice has been shown to deliver a functional DMD gene to cells lacking dystrophin protein and has been shown to increase exercise capacity[39].While promising, this technique is challenging due to the very large size of the dystrophin gene and the widespread distribution of muscles[40].
EPlLEPSY & SElZURES
Dravet syndrome
Dravet syndrome (DS), first described by Charlotte Dravet in 1978, is an early-onset epileptic syndrome characterized by a variety of refractory seizures and neurodevelopmental impairment that often persist into adulthood[41,42].The clinical features of DS typically progress over time, most commonly first presenting as a bilateral tonic-clonic in the first year of life, with half of the patients being febrile[43].The disease progresses to multiple types of seizures, often leading to poor therapeutic control and the majority of patients (93%) experiencing status epilepticus[43,44].Additionally, patients with DS will develop neurodevelopmental delay, as well as motor and cognitive impairment that will persist into adulthood.It should be noted, however, the diagnosis is highly clinical as both magnetic resonance imaging (MRI) and electroencephalogram (EEG) studies may be nonspecific[45].
Previously named severe myoclonic epilepsy of infancy, the molecular basis of DS arises from ade novomutation on chromosome 2q24 on the sodium voltage-gated channel alpha subunit (SCN1A) gene[45,46].Although the role of the SCN1A variant in the pathogenesis of DS has not been fully elucidated, some studies have suggested that the diffuse neuronal hyperactivity in DS patients is correlated with a loss of inhibitory GABAergic interneurons which have an SCN1A mutant in non-coding regions[47,48].
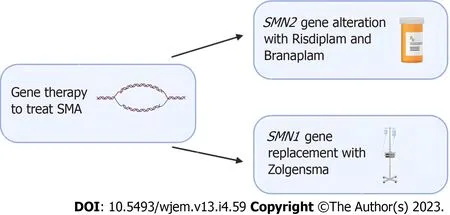
Figure 1 Emerging gene editing treatment options for spinal muscular atrophy.Risdiplam and Branaplam are oral medications which can cross the blood-brain barrier and increase the number of spinal muscular atrophy (SMA) full length proteins by targeting the SMN2 gene.For patients that require replacement of the SMA1 gene, Zolgensma is an intravenous medication that uses an adeno-associated viral vector to deliver a functional copy of the gene.Clinical trials in patients treated with Zolgensma have shown positive outcomes.SMA: Spinal muscular atrophy.
Currently, DS is managed symptomatically with a series of anti-seizure medications (valproic acid being the first line of treatment), though this regimen has variable efficacy[48].Gene-specific therapies for the causes of DS continue to gradually emerge.One study used Targeted Augmentation of Nuclear Gene Output technology, utilizing ASO, to successfully increase the expression of the SCN1A protein in mice models.In addition to a higher expression of this SCN1A product, the incidence of seizures in these DS mice was significantly reduced[49].The use of viral vectors to target genes has shown success in some diseases but poses a challenge in DS[50].The SCN1A coding sequence is 6kb long, which exceeds the carrying capacity of adeno-associated viruses[50].However, the use of a different viral vector,such as lentiviruses, poses another challenge as it demonstrates a limited spread in neural tissue therefore it cannot effectively treat large brain areas[50].Another approach to upregulating the expression of SCN1A is through the use of dCas9-based gene activation systems.One study found that the use of dCas9 resulted in the upregulation of SCN1A in brain tissue and cultured neurons[51].Overall, there have been significant developments in viral vectors and molecular techniques that demonstrate promising results.
Ohtahara syndrome
Ohtahara syndrome, also referred to as Early Infantile Developmental and Epileptic Encephalopathy (EIDEE) syndrome,is a group of devastating pediatric conditions characterized by frequent spasms in neonates and infants leading to severe cognitive and physical disabilities and even death[52,53].There are numerous causes of Ohtahara syndrome, including structural brain defects, metabolic derangements, and genetic variants.Multiple studies have identified the most common variants associated with Ohtahara syndrome are PRRT2, SCN1A, KCNQ2, and SLC2A1[54].The extensive distribution of these channelopathies and their various penetrance help explain the evolution of focal seizures to status epilepticus seen in EIDEE[54,55].
Seizure episodes in Ohtahara syndrome are initially treated with anti-seizure medications but are usually only in managing the frequency of seizures.Patients with Ohtahara syndrome with KCNQ2, SCN2A, or SCN8A mutations have been found to respond to sodium channel anti-seizure medications, such as carbamazepine, lacosamide, or phenytoin[56,57].Additionally, implementing a ketogenic diet has been shown to provide some improvement in many infants[58].Currently, the rise in genetic testing has allowed identifying the monogenic etiology of various EIDEE.An AAV-based gene replacement therapy has been proposed for Ohtahara syndrome particularly for the transmembrane sodium channel SLD13A5, though these principles could provide insight into future therapeutic targets of Ohtahara syndrome[59].However, challenges exist for gene therapy due to the limited knowledge of the disease mechanism and progression of Ohtahara syndrome.Further research is needed in patients with Ohtahara syndrome to evaluate the efficacy of gene therapies such as viral vectors.A summary can be found in Figure 2 below.
Lennox-gastaut syndrome
Lennox-gastaut syndrome (LGS) is another severe pediatric epileptic and encephalopathic disorder characterized by severe pediatric seizures, treatment-resistant epilepsy, and cognitive impairments[60,61].As in various other childhood epilepsy disorders, LGS is caused by several etiologies, including genetic predispositions, anatomical brain abnormalities,hypoxic-reperfusion encephalopathies, meningitis, and head trauma[61].The majority of children affected by LGS have underlying genetic disorders, often chromosomal syndromes, orde novopathogenic variants[61,62].
As LGS is characterized by seizures resistant to pharmacologic therapy, management often includes a multidisciplinary approach, often including dietary and surgical interventions.In one case series, 50% of children received a greater than 50% reduction in seizure frequency, and almost a quarter of the children achieved a greater than 90% reduction[63].Additionally, the serotonergic agent fenfluramine is commonly used in LGS and has been shown to have significant benefits for the reduction of generalized tonic-clonic seizures and drop seizures[64].Fenfluramine increases the level of serotonin in the extracellular compartment and acts as a serotonergic 5-HT2 receptor agonist and an alpha-1 receptor antagonist to decrease anti-epileptic activity.Since LGS seizures evolve over a patient’s life, an LGS algorithm detailing several anti-epileptics medications as well as adjunctive therapy including a ketogenic diet, possible surgical resection,and close EEG monitoring[65].
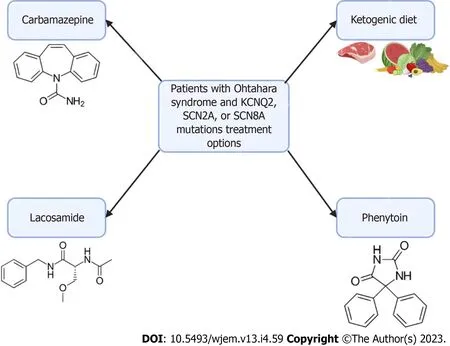
Figure 2 Treatment options for patients with Ohtahara syndrome.In patients with Ohtahara Syndrome with KCNQ2, SCN2A, or SCN8A mutations, there are several treatment options available.Carbamazepine, lacosamide, and phenytoin are sodium channel anti-seizure medications.These medications don’t offer a cure and are currently only used to manage the frequency of seizures.Additionally, implementation of a ketogenic diet has been shown to reduce seizure frequency in infants
NEURODEGENERATlVE MOVEMENT DlSORDER
Friedreich ataxia
In the discourse of neurologic orphan diseases, one important consideration in the context of neurodegenerative movement disorders is Friedreich Ataxia (FRDA).In the second half of the 19thcentury, its original description by German professor of medicine at Heidelberg, Nikolaus Friedreich, remarked FRDA was a degenerative atrophy of the posterior spinal cord columns[66].Further, between the years of 1863-1877, Friedreich published the earliest and most extensive works on “Friedreich’s ataxia”, which provided insightful descriptions of this neurological disorder as being marked with the principal abnormality of axonal thinning without axonal loss of the dorsal spinal roots[66].Unfortunately, Friedreich received little recognition for his academic effort during his lifetime, however, an anonymous obituary of “perhaps his most important work” details Friedreich’s rapid scientific progress and 59 publications[67].Nonetheless,the full extent of FRDA’s etiology couldn’t be fully appreciated until direct genetic testing became available in the late 1960s[68].Thus, the proceeding discussion will review current knowledge on this debilitating condition and evaluate the recent developments in treatment approaches.
As the most common inherited ataxia—defined as the compromised coordination of voluntary muscle movement—FRDA is an autosomal recessive, neurodegenerative disorder involving multiple organ systems including the central and peripheral nervous systems and the cardiovascular system[68,69].At the molecular level, a gene, X25, encodes a 210-amino acid protein known as “frataxin”, or FXN, and has been identified as a critical region susceptible to mutation for the FRDA locus on chromosome 9q13[69].Specifically, the most common defect leading to the development of FRDA is due to a large, homozygous intronic expansion of guanine-adenine-adenine (GAA) trinucleotide repeats in intron 1 of the said frataxin gene[70,71].Frataxin, a highly conserved small mitochondrial protein, is required for efficient regulation and homeostasis of cellular iron stores[72].Consequently, the dysregulation of this critical molecule’s function is associated with mitochondrial iron overload, iron-sulfur cluster biosynthesis, and free radical oxidative stress[73,74].A graphical representation of the molecular etiology and subsequent biological consequences of dysfunctional frataxin protein function can be seen in Figure 3 below.
Clinically, due to frataxin’s ubiquitous biochemical role in numerous cellular pathways, humans with frataxin deficiency manifest with dysfunction in the central and peripheral nervous system, heart, skeleton, and even endocrine organs including the pancreas[71].Regarding FRDA in particular, many of these patients classically present with neuropathologic disabilities including progressive ataxia, peripheral sensory loss, and muscle weakness beginning between the ages of 5 and 15 years old[76].The aforementioned manifestations are chiefly secondary to neuropathy in the dorsal root ganglia, accompanied by degeneration of both peripheral sensory nerve fibers and posterior columns of the spinal cord[76].Moreover, non-neurological areas of morbidity include the heart, typically in the form of left ventricular hypertrophy due to mitochondrial proliferation, and the pancreas, with approximately 10% of all FRDA patients developing diabetes mellitus[76,77].
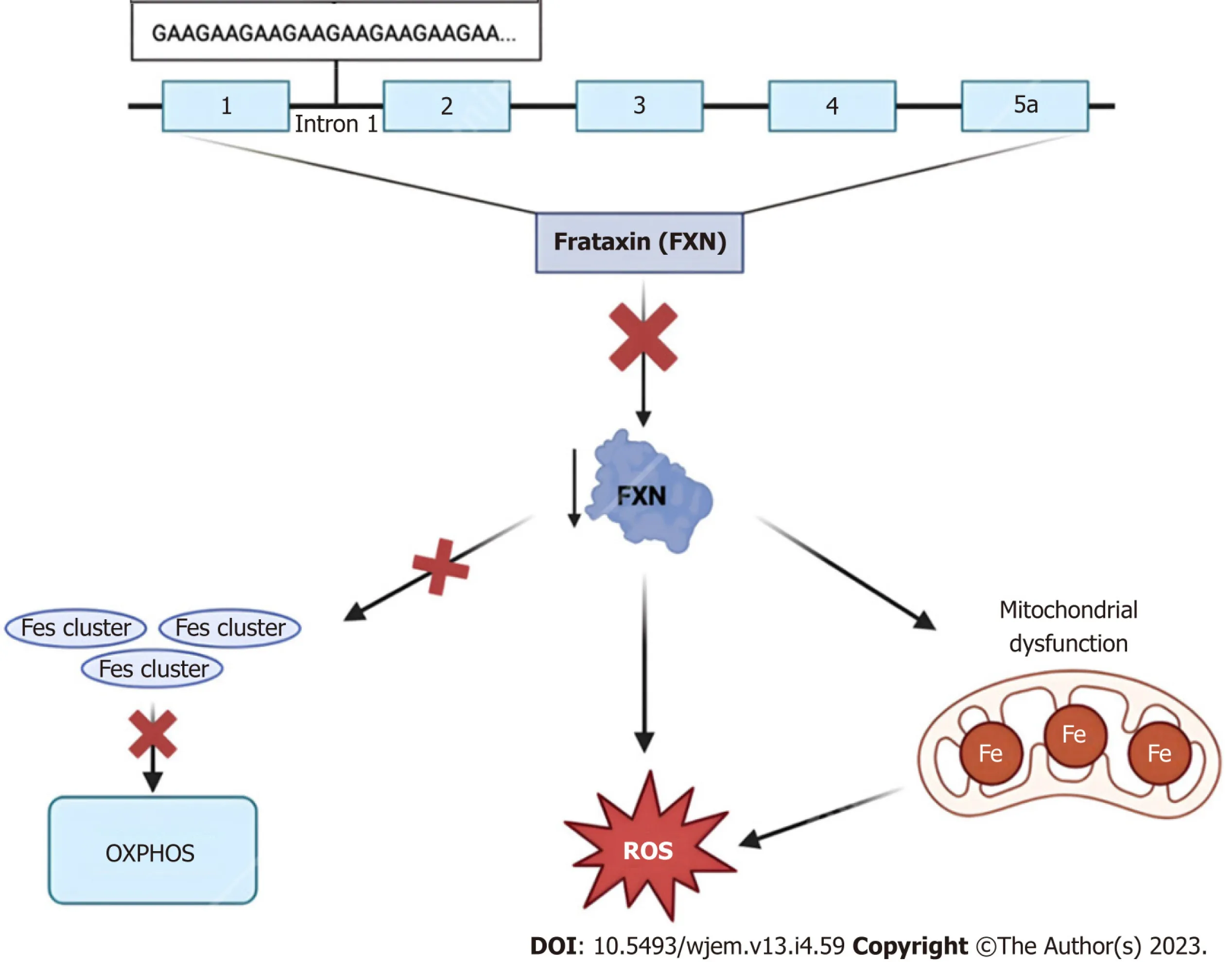
Figure 3 Frataxin protein dysregulation.The 2100 amino acid protein frataxin is encoded within the first intron of the FXN gene on chromosome 9q13.Due to trinucleotide repeat expansions ranging from approximately 44-1700 “GAA” triplet sequences, affected individuals experience numerous characteristic signs and symptoms of Friedreich Ataxia.At the cellular level, dysregulation of this small mitochondrial protein results in the overproduction of iron-sulfur clusters, free radical oxidative stress, and mitochondrial iron overload[75].FXN: Frataxin; ROS: Reactive oxygen species.
Before the dawn of genetic testing, the constellation of the above-mentioned signs and symptoms as well as MRI was employed in the diagnosis of FRDA[78].Presently, however, this neurologic disorder is more precisely diagnosed using modern genetic testing such as Southern blot and conventional polymerase chain reaction (PCR) techniques[79].The accurate diagnosis of FRDA helps in differentiating it from other ataxias and provides a guide for physicians to appropriately tackle the treatment and management of this progressive disorder.Despite the heterogeneity of symptomatic presentation, most patients will lose the ability to independently walk, stand, and/or sit within 10-15 years of initial disease onset[76].Therefore, there is a persisting call for advancements in techniques and approaches for treating FRDA.
In the past, there were no effective protective pharmacological agents for FRDA, and thus treatment was aimed at symptomatic management and physical therapy for impaired motor function[80].Further, aggressive surveillance was and still is used in managing progressive cardiomyopathy, arrhythmias, and diabetes mellitus to improve the quality of life for these patients[80,81].More recently, treatment approaches to address the mitochondrial dysfunction caused by a mutation to the frataxin protein include mitochondrial function enhancers, free radical scavengers, and iron chelators such as coenzyme Q10and its synthetic analog idebenone, as well as vitamin E[82].Although the employment of such molecules is mechanistically sound, the pre-clinical and clinical trial data on their use has demonstrated little to no therapeutic effect across multiple studies and even worsened enzymatic activity[80].As of February 2023, however, the first FDA-approved treatment for FRDA became available within the United States.Developed by Reata Pharmaceuticals,Inc., the small molecule Omaveloxolone, or SKYCLARYS™, is an orally active drug aimed at alleviating oxidative stress,mitochondrial damage and dysfunction as a direct result of Nrf2 pathway suppression found in Friedreich patients[83].As a novel pharmaceutical with early proven safety and effectiveness, Omaveloxolone is a promising emerging option for FRDA treatment.
In addition to symptomatic treatment and management, newer therapeutics for neurological disorders are being developed, evidenced by the rapid increase in research surrounding gene and cell therapy for FRDA between the years 2000 and the present day.Considering the severity of neurophysiological abnormalities is strongly correlated with the size of the GAA repeat expansion implicated in the genetic inheritance of FRDA, many of these novel techniques are aimed at altering this pattern.Specifically, the use of the CRISPR-Cas9 system in GAA expansion-based animal models such as YG8-derived cells and mouse models demonstrated promising results in the successful editing of GAA expansion bothin vitroandin vivo[84].Contrastingly, instead of altering the primary genetic mutation, other more recent methods of ataxia reversal are aimed at inducing the expression of the frataxin protein itself[85].Particularly, Piguetet al[86]demonstrated the complete reversal of sensory ataxia and cardiomyopathyviaviral vector-based introduction of AAVexpressing frataxin (AAV-FXN) in cells and mouse models.Finally, other prospective therapeutics look to stimulate the direct transcription of FXN and/or increase FXN mRNA stabilityviainterferon administration, or indirectlyviathe stimulation of NRF2—a transcription factor whose levels are intimately associated with FXN mRNA expression[87].Altogether, these animal-based models hold promising treatment options for future application to clinical medicine, for now, more research is necessary before translation into FRDA human-based clinical trials.
lNHlBlTED COGNlTlVE DEVELOPMENT
Prader-Willi syndrome (PWS) and Angelman syndrome (AS) are the first known examples of diseases relative to imprinted genes.These diseases have their own clinically distinct behavioral, cognitive, and neurologic phenotypes.However, they are still considered “sister disorders” due to the common origin of the disease from the imprint gene abnormalities in the 15q11-q13 region[88].The difference in the diseases is that with PWS, the contribution is paternal whereas with AS, the contribution is maternal.This parent-of-origin difference is the reason for the variable expression of the disease depending on the sex of the parent from whom the disease is inherited[88,89].
PWS
PWS is suspected in individuals that present with clinical features of hypotonia (during the first few years) that lead to hyperphagia, hypogonadism, short stature, and mild mental retardation[90].However, regarding the 15q11-q13 region,the abnormality is suggestive of PWS, but not diagnostic.The diagnosis of PWS is established by identifying the abnormal DNA-methylation and maternal-allele imprinting in the Prader-Willi critical region.This can be due to either the deletion of the paternal allele, maternal disomy, and/or imprinting defect causing the absence of the paternal allele[91].Moreover, point mutation does not cause PWS because it is a factor of multiple gene products.An exception to this rule is when the point mutation is seen in the imprinting control region[92].In these cases, although very rare, the loss of function has been seen to contribute to many different aspects of the PWS phenotype[93].
The underlying molecular defect in PWS further opens a great scope of molecular treatment and epigenetic therapy.An increasing number of studies are pointing toward gene SNORD116 being the main causative player in PWS[94-96].Deactivation of the SNORD116 gene is associated with a complex of a zinc finger protein ZNF274 and SETDB1 a histone H3 methyltransferase[97-99].Thus, CRISPR-Cas systems can be employed to deactivate ZNF274 and/or SETDB1 and increase the expression of SNORD116.One of the drawbacks to this approach is that the SETDB1 is not specific and thus,targeting it can have many off-label systemic effects.
Another approach explored in many studies was to inhibit G9a, another histone H3 methyltransferase, which restored the targeted genes from the silenced maternal chromosome[100-102].Based on these studies, the use of a G9a inhibitor improved perinatal lethality and poor growth, which ultimately can improve the life span[100-102].But again, one of the drawbacks of these research studies is that they are all performed with either mouse models or PWS patient-derived cells.Thus, the efficacy of gene therapy in PWS patients is yet to be explored and proven to be beneficial.Moreover, during the development of new drug therapies, focus should be put on off-target effects.Epigenetic drugs are known to have complex and broad effects, and thus, the development of specific molecular therapy for PWS patients is necessary.
AS
AS clinically presents as severe developmental delay, minimal or no speech, difficulty walking, and a unique behavioral phenotype that includes frequent smiling and excessive laughter[103].As mentioned above, defects in the 15q11.2-q13 chromosome region are the genetic basis of both PWS and AS.In terms of AS however, the maternally expressed allele does not produce functional gene product, and the paternal allele is imprinted.The gene of interest in terms of AS is the UBE3A sequence which is mutated in certain individuals with AS[103].
In terms of epigenetic therapy, restoration of the mutated UBE3A gene expression is more favorable and plausible rather than targeting known activities of the molecule.However, there haven’t been any successful ways researched to do so.Thus, expressing the silenced paternal UBE3A has been an adequate alternative.The most common way proposed is by administering topoisomerase-I inhibitors and reactivating the inhibition of paternal UBE3A.
Topoisomerase-I inhibitors like topotecan and irinotecan are FDA-approved chemotherapeutic drugs.Moreover, these drugs are also shown to promote the expression of paternal UBE3A in a dose-dependent manner.The drugs work by reactivating the geneviareduced transcription of an imprinted antisense RNA[104-106].Despite proving their potential,topoisomerase inhibitors are not currently used as a first-line treatment for AS due to their side effects.According to one study, topoisomerase inhibitors also reduced the levels of other genes that are linked to autism, thus increasing the potential of autism characteristics in individuals[107].
Thus, as discussed, all the approved therapies for PWS and AS target the management of symptoms rather than the actual disease.Thus, there is currently no cure for either AS or PWS.Gene and molecular therapies offer a promising way into precision medicine and the development of novel therapies however a lot more research needs to be done regarding offsetting the systemic side effects.A summary of the chromosome 15 abnormalities in PWS and AS is shown in Figure 4 below.
NEURON DETERlORATlON
Amyotrophic lateral sclerosis
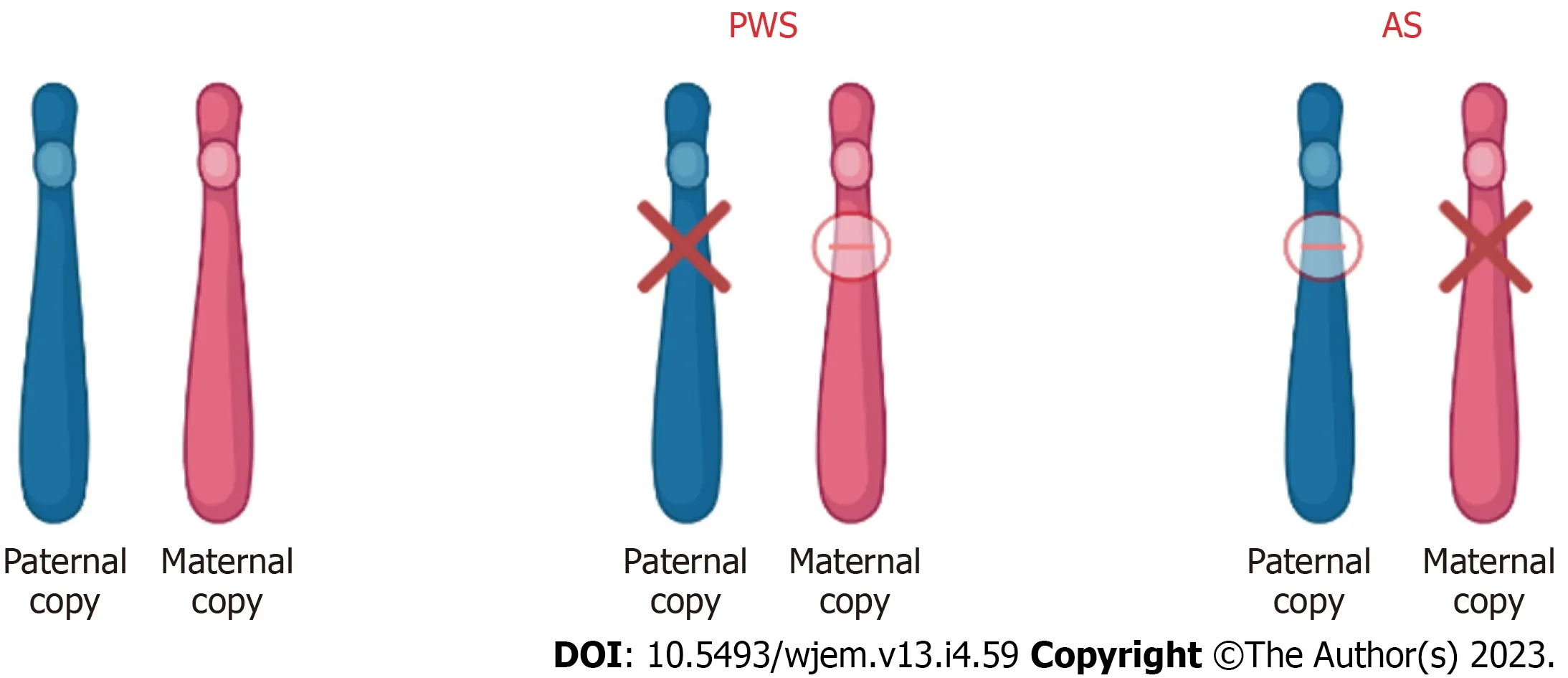
Figure 4 Chromosome 15 (15q11-13) abnormality in patients with Prader-Willi syndrome and Angelman syndrome.In patients with Prader-Willi syndrome (PWS) and Angelman syndrome (AS), the imprinted gene abnormality is the 15q11-q13 region of chromosome 15.Although the abnormality is in the same region, the difference in disease and phenotype depends on the parental contribution.In PWS, the disease results due to loss of paternal gene expression.In AS, the disease results due to loss of maternal gene expression.PWS: Prader-Willi syndrome; AS: Angelman syndrome.
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder affecting both the upper and lower motor neurons and is the most common motor neuron disease in adults[108-110].The onset of ALS symptoms typically occurs between 50-70 years old and is more common in males than females[111-113].While some individuals can survive over 10 years with ALS, most survive less than 3 years due to the rapid disease progression[114].Current treatments focus on the treatment of symptoms; however, their effect is often minimal by extending a patient’s survival by only three months[112].Although the etiology of ALS is currently unknown, there does appear to be a genetic component as nearly 10 percent of diagnosed individuals reported having a family member with ALS and are said to have familial ALS (fALS),while the remaining individuals are classified as having sporadic ALS (sALS)[115].
In recent years, the number of genetic mutations linked to ALS has increased.Superoxide dismutase-1 (SOD1) was the first gene discovered to be associated with ALS, and mutations of the SOD1 gene account for 10%-15% of fALS and 1%-2% of sALS[110,115-117].More recently, mutations to the C90RF72 gene have been associated with 30%-40% of fALS and nearly 6% of sALS[115,116].Mutations to other genes such as TDP-43, FUS, OPTN, TBK, GRN, NEK1, and C21ORF2 have also been linked to ALS[115,116].Current evidence suggests that many of these mutations result in the aggregation of misfolded proteins, which then causes ALS[110,112].Furthermore, knockout models tested on mice have shown that the removal of these genes typically does not result in symptoms of ALS, suggesting that these mutations result in a gain of function[112,116,117].
Current ALS treatments in clinical trials focus on gene therapy as opposed to treatment of the symptoms of ALS.These treatments attempt to replace the mutated gene, inactivate the mutated gene, or introduce a new gene to fight ALS[117].Tofersen, an ASO, is currently in clinical trials for individuals suffering from ALS because of a SOD1 mutation.Tofersen causes the degradation of SOD1 mRNA, decreasing the amount of SOD1 protein[118].However, ASOs such as Tofersen are unable to cross the blood-brain barrier and must be injected into the cerebrospinal fluid[117,118].This can cause many unwanted side effects, such as inflammation, infection, and long and repeated injections[117].As a result, nanoparticles,such as calcium phosphate lipid nanoparticles, are being developed to reduce the need for direct injection of ASOs into the CSF[117].Additional research is being conducted on AAV vectors for the modification of genes.The major advantage of AAV vectors is that only one injection is necessary[116].AAV vectors have the potential to deliver gene-silencing material, such as antisense sequences, to mutated genes such as the SOD1 gene[116].However, immunoreactivity to AAV has been documented in both animal and human studies presenting future challenges to the use of AAV vectors for ALS treatment[116].
Huntington’s disease
Huntington’s disease (HD) is a neurodegenerative disorder resulting from the expansion of a CAG trinucleotide sequence on theHTTgene which encodes the huntingtin protein[119-121].For an individual to be at risk for HD, they must have greater than 36 glutamine repeats, and as the number of repeats increases the age of onset typically decreases[120].HD is inherited in an autosomal dominant manner, with symptoms first appearing in mid-life[121,122].Individuals with HD experience cognitive, motor, and psychiatric symptoms that are progressive over time[119].Current treatments for HD focus on treating symptoms but often have limited benefits, and there is no available disease-modifying treatment[121,123].
The HTT gene encodes the huntingtin protein, which is involved in a variety of cellular functions including cell division, vesicle transportation, and transcription regulation[120].Repeat CAG trinucleotide sequences found on exon 1 of the HTT gene cause the formation of mutant huntingtin protein aggregates within neurons[124,125].Individuals who possess 60 or more CAG repeats will develop HD before the age of 20[124,125].However, the CAG repeat length explains less than 50% of the HD age of onset[124].Other factors such as glutamic acid polymorphisms and glutamate and Nmethyl-D-aspartate receptor polymorphisms also play a role in the onset of HD[124].These genetic modifiers all present potential targets for genetic treatment for HD.
As a result of the many functions of the huntingtin protein, the embryonic knockout of theHTTgene in mice was lethal[120,121].Furthermore, the inactivation of the HTT gene after birth has been linked to neurological deterioration in mice[121].Because of this, many HD disease-modifying treatments being researched focus on gene editing as opposed to gene silencing[125].In a phase 1-2a clinical trial conducted between 2015-2017, an ASO known as IONIS-HTTRxwas studied for its safety, ability to remain in the CSF, and ability to reduce mutant HTT in the CSF[126].Upon completion of this trial, it was found that IONIS-HTTRxdid not increase the number of adverse effects and resulted in a dose-dependent reduction of the mutant HTT concentration[126].However, phase three trials show that IONIS-HTTRxmay cause worse motor and cognitive function[127].Along with ASOs, RNA interference (RNAi) technology is being developed to treat HD.RNAi technology attempts to decrease the amount of mutant HTT being translated using small non-coding RNAs[127].This RNAi technology is typically delivered through an AAV.However, this technology is in the early stages of development,with significant testing still needed.
TUMORS
Neurofibromatosis-1
Neurofibromatosis-1 (NF-1), also known as Von Recklinghausen disease, is an autosomal dominant disorder characterized by changes in the nervous system, bones, and skin[128].Risks associated with NF-1 include bone abnormalities,vasculopathy, and cognitive impairment[128].NF-1 is also the common type of hamartoma neoplastic syndrome, which is the formation of benign tumorlike malformations consisting of abnormal cells and tissues[128].Neurofibromatosis also consists of neurofibromatosis type 2 (NF-2) and Schwannmatosis[128].NF-2 presents with similar cutaneous manifestations as NF-1 but mainly exhibits schwannomas (tumors of the nervous system), meningiomas (tumors of the meninges),and ependymomas (brain and spinal cord tumors)[129].Of the three kinds of NF, NF-1 accounts for 96% of all cases, NF-2 accounts for 3% and Schwannomatosis accounts for less than 1%[130].
NF-1 is known as an autosomal dominant disorder that affects 1 in 2600 to 3000 individuals.All generations are included.The expression of this disease differs between individuals of the same family and from one affected family to another[128].The NF-1 gene is located on chromosome 17 and is a tumor suppressor gene.The NF-1 gene produces a product called neurofibromin, which catalyzes the hydrolysis of guanosine triphosphate (GTP)-bound Ras to guanosine diphosphate-bound Ras, ultimately inactivating Ras GTPase[131].Neurofibromin is expressed in various tissues throughout the body and the function of NF-1 is to modulate the activity of the RAS pathway[132].This pathway in turn delivers signals from the granulocyte-monocyte colony-stimulating factor to proliferating cells[132].In turn, the NF-1 protein promotes the conversion of the activated complex, Ras-GTP, to Ras-GDP, the inactivated form[133].When the NF-1 gene is mutated or deleted, the typical phenotype of NF-1 results due to activation of the Ras-GTP, ultimately resulting in cell growth and prolferation[128].With this disorder, penetrance is complete[134].NF-1 has a high degree of variability in clinical presentation, which may include cutaneous, bony, vascular, and cognitive features along with multiple neoplasms.Due to these manifestations overlapping with other genetic conditions, accurate diagnosis of NF1 is important for clinical care and genetic counseling[135].The mechanism of neurofibromatosis-1 is summarized in Figure 5 below.
New gene techniques were shown to help with NF-1-related pain.A study conducted on this rare autosomal disease suggested that collapsing response mediator protein 2 (CRMP2) is a key target for therapeutic intervention[136].Moutalet al[136] highlighted there is a direct connection between the amount of neurofibromin expressed and pain.CRMP2 regulates the activity of calcium channels and increases the Ca2+current and release in sensory neurons[137].Additionally, CRMP2 binds to the C-terminus of neurofibromin, suggesting a possible correlation between CRMP2 and the pain experienced by patients with NF-1[136].In the study, they used clustered regularly interspaced short palindromic repeats of the CRISPR-associated 9 (CAS9 genome), a commonly used DNA editing device[136].With this,the researchers created a novel rat model of NF-1-related pain[136].The delivery guide of Cas9 nuclease plasmid was used to generate allele-specific C-terminal truncation of neurofibromin[136].Additionally, researchers used (S)-LCM, an inactive enantiomer of the drug Vimpat to inhibit CRMP2 phosphorylation, uncoupling CRMP2 from the NF-1 protein[136].The rats with truncation of neurofibromin showed increases in voltage-gated calcium and sodium resulting in increased nociceptor excitability and behavioral hyperalgesia[136].As the protein CRMP2 regulates these channels and binds to the C-terminus of neurofibromin, this indicated a possible mechanism underlying NF1 pain[136].Targeting CRMP2 phosphorylation offers a new therapeutic way to manage the pain associated with NF-1.However, future research is needed in human trials to further investigate the link between CRMP2 and the NF-1 protein.
In children with neurofibromatosis 1, low-grade gliomas are the most common tumor and often result in significant visual loss due to tumor progression along with motor deficits[138].The current therapeutical agent used for the management of tumor progression is vincristine/carboplatin, or vinblastine, a chemotherapeutic agent[139].Although this agent prevents tumor progression, restoration of vision or motor function is rare[140].A new type of therapeutic approach involves the use of mTOR inhibitors[140].When mTOR is activated, abnormal cell growth results leading to cell proliferation and the formation of new blood vessels through angiogenesis[140].Everolimus is derived from rapamycin and inhibits angiogenesis, hypoxia-inducible factor 1, and vascular endothelial growth factor production, and ultimately prevents the proliferation of cells[141].When Everolimus was given to patients with NF-1, the tumor progression was significantly halted and demonstrated sufficient penetration of the blood-brain barrier[141].This offers a promising therapeutic for children with low-grade gliomas as it is available orally and has minimal toxicities.However, further researcher is needed on Everolimus in combination with other agents to determine if it is the superior therapeutic agent or if a combination offers better results.
Tuberous sclerosis
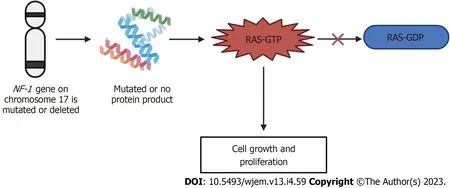
Figure 5 Mechanism of neurofibromatosis-1.If the neurofibromatosis-1 gene on chromosome 17 is either mutated or deleted, either no protein product is formed, or a mutated protein is made.This mutation or deletion then results in the inhibition of the conversion of Ras-GTP to Ras-GDP, ultimately resulting in the increased signaling of Ras-GTP and leading to cell growth and proliferation, producing the neurofibromatosis phenotype.GTP: Guanosine triphosphate; GDP:Guanosine 5'-diphosphate; NF-1: Neurofibromatosis-1.
Tuberous sclerosis is an inherited autosomal dominant neurocutaneous genetic disorder[142].This genetic disorder affects multiple systems and often presents in children with skin lesions, seizures, and hamartomas of the brain, kidney,and heart[142].Additionally, tuberous sclerosis impacts 1 in 5500 individuals.Affected individuals may also present with developmental delays[142].Cardiac rhabdomyomas of cortical tubers may also be present prenatally[142].Adulthood signs show osseous, renal, or pulmonary lesions[142].Skin lesions are found in 90% of patients of all ages whilst,hypopigmented macules are found in early childhood[142].Ungual fibromas appear during puberty and facial angiofibromas are more commonly found towards adolescence[143].
Tuberous sclerosis disorder arises from four mutations in tuberous sclerosis complex 1 (TSC1) (9q34) and tuberous sclerosis complex 2 (TSC2) (16p13.3) genes which respectively encode hamartin and tuberin[144,145].This disorder has been known to have a broad spectrum of mutations in both genes[144].No regions seem more liable to mutations and the frequency is consistently higher in TSC2 rather than in TSC1[144].15% of patients admitted who meet clinical criteria for tuberous sclerosis demonstrated no identifiable genetic mutations[143].The disease itself is caused by a mutation of these genes and causes dysfunction of proteins hamartin or tuberin[143].Hamartin’s role helps control the proliferation of cell growth, division, and cell size[143].Tuberin functions to regulate cell growth and protein synthesis through the downstream inhibition of mTOR[146].Therefore, the loss of either of these proteins can lead to the overgrowth of lesions in many vital organs[146].
Gene therapy for tuberous sclerosis type 2 was conducted on a mouse model by delivering an adeno-associated virus(AAv9) which encoded a condensed form of tuberin[147].A mouse model of TSC2 was generated by AAV-Cre recombinase disruption of Tsc2-floxed alleles at birth, leading to a shortened lifespan (mean 58 d) and brain pathology consistent with TSC[147].When these mice were injected intravenously on day 21 with AAV9-cTuberin, the mean survival was extended to 462 d with a reduction in brain pathology[147].This study demonstrated the potential of treating life-threatening TSC2 Lesions with a single intravenous injection of AAV9-cTuberin[147].Preventions for this disease will vary depending on the developmental stage of the specific individual as tuberous sclerosis has a highly variable clinical course and the prognosis may be uncertain[147].Options pertaining to tuberous sclerosis are limited to surgery for treating symptoms of tuberous sclerosis related to the growth of hamartomas[148].Therefore, to better understand the genetic causation for this disease, clinical trials of mammalian target of rapamycin inhibitors, including sirolimus and Everolimus have been conducted[149].mTOR is an evolutionary conserved serine-threonine kinase that regulates cell growth and cell survival[149].The connection between TSC and mTOR led to the clinical use of mTOR allosteric inhibitors, Sirolimus and Everolimus[149].Both sirolimus and Everolimus inhibit mTOR selectively with similar molecular mechanisms but distinct clinical profiles[149].Everolimus has been approved for subependymal giant cell astrocytomas and renal angiomyolipomas in TSC patients[149].However, sirolimus is not approved for TSC and has undergone considerable investigation to treat various aspects of TSC.However, the use of sirolimus has been studied in older children and adults with TSC[150-153].One study published in 2023 investigated the use of sirolimus in children under the age of 2 with tuberous sclerosis complex and found that sirolimus was safe to use in children[154].The study reported that the most common adverse events due to sirolimus use included anemia, hyperlipidemia, and thrombocytosis, which were able to be managed well[154].Despite proving its safety through this study, further research is needed into sirolimus to demonstrate safety and efficacy through larger studies and clinical trials, but it offers a promising therapeutic option for patients with tuberous sclerosis.
CONCLUSlON
Neurologic orphan diseases impact less than 200000 individuals within the United States and because they’re rare,diagnosis and treatment are often difficult.However, in recent years, data has suggested that the use of whole genome sequencing has increased the diagnosis of neurologic orphan diseases, allowing patients who have these conditions to be more easily identified.With new advances in technology, more research is being devoted to developing therapeutic options for patients with neurologic orphan diseases.Such advances in research include the use of AAV vectors which have shown positive results in SMA, ALS, Friedreich Ataxia, and NF-1.Additionally, the use of CRISPR/Cas has demonstrated promising results in DMD, Dravet syndrome, Friedreich Ataxia, NF-1, and Tuberous Sclerosis.The use of therapeutics that can cross the blood-brain barrier, such as Risdiplam and Branaplam, has increased the number of treatment options available for patients with SMA.Additionally, research into mTOR inhibitors offers a promising option for patients with NF-1 and tuberous sclerosis.Although further research is needed, these treatment options can significantly impact the quality of life and survival of patients with neurologic orphan diseases.
FOOTNOTES
Author contributions:Lucke-Wold B contributed to conceptualization and supervision; Kioutchoukova I, Foster D, Thakkar R, Foreman M, Burgess B, Toms R, Valero EM contributed to writing-original draft presentation; Ivelina Kioutchoukova, Devon Foster contributed to writing-editing and review.
Conflict-of-interest statement:All the authors declare no conflict of interest.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial.See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:United States
ORClD number:Ivelina P Kioutchoukova 0009-0001-7467-2382; Devon T Foster 0009-0007-9411-9051; Rajvi N Thakkar 0009-0003-2375-5078;Marco A Foreman 0000-0002-5551-7061; Brandon J Burgess 0009-0008-7203-8469; Rebecca M Toms 0009-0005-5539-9754; Eduardo E Molina Valero 0000-0002-3235-4079; Brandon Lucke-Wold 0000-0001-6577-4080.
S-Editor:Liu JH
L-Editor:A
P-Editor: Xu ZH
