Overview of angiogenesis and oxidative stress in cancer
2023-11-18LuigiGaetanoAndrioloVittoriaCammisottoAlesandraSpagnoliDaniloAlunniFegatelliMicheleChiconeGaetanoDiRienzoVladimiroDellAnnaGiambattistaLobreglioGiovanniSerioPasqualePignatelli
Luigi Gaetano Andriolo, Vittoria Cammisotto, Alesandra Spagnoli, Danilo Alunni Fegatelli, Michele Chicone,Gaetano Di Rienzo, Vladimiro Dell'Anna, Giambattista Lobreglio, Giovanni Serio, Pasquale Pignatelli
Abstract Neoplasms can be considered as a group of aberrant cells that need more vascular supply to fulfill all their functions.Therefore, they promote angiogenesis through the same neovascularization pathway used physiologically.Angiogenesis is a process characterized by a heterogeneous distribution of oxygen caused by the tumor and oxidative stress; the latter being one of the most powerful stimuli of angiogenesis.As a result of altered tumor metabolism due to hypoxia, acidosis occurs.The angiogenic process and oxidative stress can be detected by measuring serum and tissue biomarkers.The study of the mechanisms underlying angiogenesis and oxidative stress could lead to the identification of new biomarkers,ameliorating the selection of patients with neoplasms and the prediction of their response to possible anti-tumor therapies.In particular, in the treatment of patients with similar clinical tumor phenotypes but different prognoses, the new biomarkers could be useful.Moreover, they may lead to a better understanding of the mechanisms underlying drug resistance.Experimental studies show that blocking the vascular supply results in antiproliferative activity in vivo in neuroendocrine tumor cells, which require a high vascular supply.
Key Words: Neuroendocrine lung tumors; Angiogenesis; Oxidative stress; Neuroendocrine serum markers; Neuroendocrine tissue markers; Future therapy
INTRODUCTION
The angiogenesis process consists of the generation of new blood vessels.The migration and proliferation of endothelial cells from already existing vessels to new vessels are crucial in this process.During embryonic development, these cells are particularly active, whereas in the adult their turnover is slow and limited to certain physiological phenomena, such as ovulation, tissue repair, and scarring processes[1].
Angiogenesis is the result of a well-balanced process between proangiogenic and antiangiogenic factors.This balance can fail due to specific stimuli such as hypoxia, creating a pathological angiogenic process[2].The prevalence of proangiogenic factors is associated with serious diseases, such as cancer, and with inflammatory and degenerative diseases, such as retinopathies, rheumatoid arthritis, and psoriasis.Insufficient angiogenesis is the basis of obliterating vascular diseases, such as obstructive coronary artery disease or peripheral obstructive arterial disease (Buerger’s disease), which are characterized by the downstream tissue ischemia of vascular occlusions[3].
Neoplasms can be considered complex biological structures constituted by aberrant cells and endowed with specific functions; there are mesenchymal-derived cells, inflammatory cells, and vascular cells communicating with one another[4].To fulfill all their functions, including growth and metastasis, they can promote angiogenesis through the same neovascularization pathway used physiologically.Tumor progression occurs due to the proliferation of the tumor cells themselves and the interactions that the neoplasm sets up within the tumor microenvironment where distinct types of tumor cells secrete key cytokines[5] for tumor progression and metastasis[6].
Cancer cells in active and continuous replication need a constant supply of oxygen and nutrients.For this reason, the first mechanism that cancer cells use to ensure the survival and growth of its cells is angiogenesis.However, neoplastic angiogenesis is an aberrant process associated with the formation of tortuous vessels that are insufficient to fulfill cellular needs.Acidosis is the consequence of altered tumor metabolism in response to hypoxia and the heterogeneous distribution of oxygen between the core and periphery that tumor angiogenesis helps to create.In this way, the acidic environment selects a more aggressive neoplastic cell phenotype with a greater invasive and metastatic phenotype.
Metabolic, hypoxic, and oxidative stress is considered a distinctive marker of cancer[7].To survive the metabolic stresses, cancer cells activate different types of mechanisms including evasion of apoptosis and immune surveillance,increasing the angiogenic activity to enhance the provision of oxygen and nutrients, activation of the epithelialmesenchymal transition (EMT), and metastasis[7,8].Positive feedback between angiogenesis and oxidative stress is evident when a cellular mechanism stands for both the stimulus and the result of this process (Figure 1).
Tumor-induced angiogenesis begins with the release and activation of many growth factors[9].The most important of which is vascular endothelial growth factor (VEGF) with its receptors.The mechanism of angiogenesis is complex, and it passes through stages well defined by changes in the endothelium and the extracellular matrix[10].It can be schematically described as follows.The first stage of angiogenesis is characterized by the “destabilization” of pre-existing vessels and the loss of connection between endothelial cells due to increased vascular permeability.The proliferation phase of the endothelial cells follows with the formation of new vessels.Various proteolytic enzymes are released during these phases and alter the density of the extracellular matrix to help the migratory activity of endothelial cells.The third stage of angiogenesis is characterized by the formation of primitive capillaries.Finally, the last stage involves the recruitment of supportive periendothelial cells, such as pericytes and muscle cells, as well as the reorganization of periendothelial cells[11].
The most powerful stimulus for angiogenesis is hypoxia.Hypoxia and angiogenic factors released by the tumor destabilize the pericytes and stimulate continuous angiogenesis[12].Tumors maintain hypoxia primarily due to the heterogeneous distribution of oxygen between the core and the periphery that cancer cells generate[13]; this situation is also associated with acidosis.By maintaining a low pH, cancer cells can evade immune cells and be chemoresistant[14].
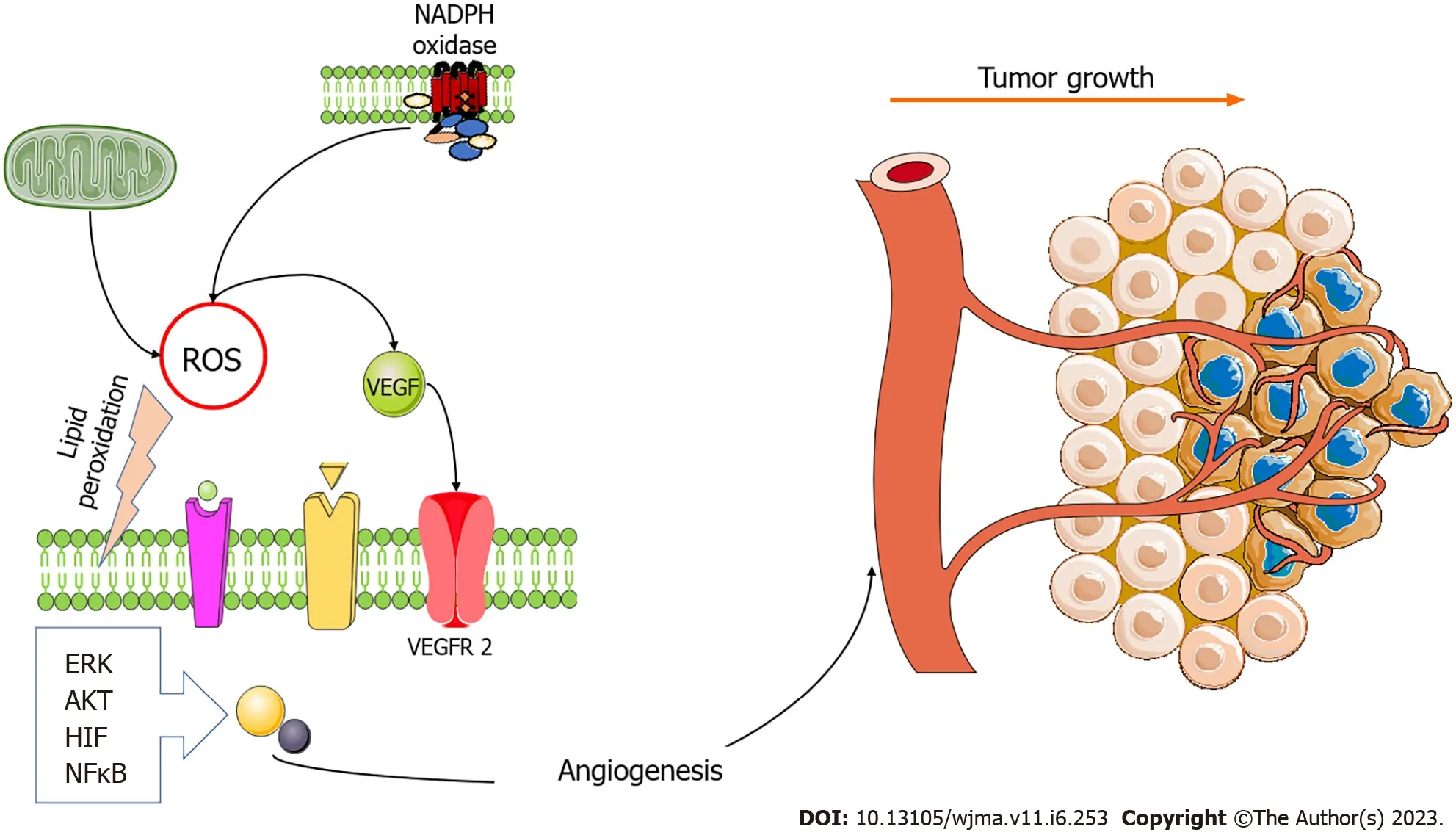
Figure 1 The two main sources of oxidative stress, mitochondria, and nicotinamide adenine dinucleotide oxidases generate reactive oxygen species that trigger angiogenesis.The vascular endothelial growth factor (VEGF) pathway is modulated by reactive oxygen species (ROS), and oxidative stress stimulates VEGF production in several cell types, including endothelial cells.ROS enhance angiogenesis by increasing hypoxia-inducible factor (HIF)1 α, protein kinase B (AKT), and regulated extracellular kinase (ERK).However, oxidative stress also induces angiogenesis in a VEGF-independent manner by lipid peroxidation and generating metabolites that act either as ligands or by inducing post-translational modifications of proteins within angiogenic signaling pathways,such as nuclear factor kappa-light-chain enhancer of activated B cells (NFκB) activation pathways.Figure was prepared using images from Servier Medical Art by Servier (https://smart.servier.com), which are licensed under a Creative Commons Attribution 3.0 Unsupported License.NADPH: Nicotinamide adenine dinucleotide;VEGFR2: Vascular endothelial growth factor receptor 2.
Reactive species, mainly represented by reactive oxygen species (ROS), are products generated by metabolic reactions that take place in the mitochondria of eukaryotic cells.If these reach a certain level they can be toxic to the cells.Physiological concentrations of reactive species can generally transduce signals before they are eliminated, whereas tumor cells need high concentrations of ROS to support their high proliferation rate due to their metabolism[15].
Among the several cellular strategies adopted by tumors to develop resistance to ROS are the so-called alternative metabolic pathways.These pathways prevent the accumulation of ROS without reducing the metabolic energy required by the tumor cells.The glycolysis with its parallel pathway and the pentose phosphate pathway, are examples of these pathways.The ROS levels are a sign of the damage that cells can withstand[16].
The therapeutic implications that follow are particularly important since the radiotherapy and chemotherapy currently available conduct their antitumor action precisely through the regulation of ROS levels.Therefore, the clinical response to pro-oxidant therapies has to be considered to enable truly personalized therapies.Consequently, the discovery of biomarkers capable of predicting this response is a challenge[17].
Somatostatin is a ubiquitous polypeptide produced by the delta cells of the digestive system and is present in the intramural plexuses of the intestine.Tumors originating from these cells produce and secrete somatostatin.Somatostatin exists in two biologically active forms, namely SS-14 and SS-28[18].
Several functions of somatostatin in the central nervous system are described.These include neuromodulatory,locomotor, and cognitive functions, inhibition of basal and stimulated secretion of distinct types of endocrine and exocrine cells, and regulation of cell proliferation and differentiation[19].Specific membrane receptors are bound by somatostatin, of which there are five different subtypes called somatostatin receptors 1-5 (SSTR 1-5).These have maintained structural homology between distinct species (40%-60% of structural homologies) and mediate different biological actions by activating different intracellular signaling pathways[20,21].
Tumors that produce somatostatin have a typical histological architecture common to all neuroendocrine tumors(NETs) and a high somatostatin production.Somatostatin is a powerful inhibitor of neovascularization as many experimental data have shown.SSTR are expressed on endothelial cells, and the activation of quiescent endothelium is associated with an upregulation of SSTR2.
Somatostatin agonists inhibit VEGF, basic fibroblast growth factor, and growth hormone/insulin-like growth factor 1.Consequently, they can negatively regulate angiogenesis[22].Furthermore, somatostatin can function as a powerful antitumor agentin vivoinhibiting both endothelial nitric oxide synthase and mitogen-activated protein kinases (MAPK)through SSTR3[23].
NETs represent a neoplasm that most benefit from metabolic radiotherapy and treatment with antiangiogenesis and pro-oxidant drugs.The presence of marked vascularization is a distinctive feature in most NETs, and this characteristic can be considered one of the diagnostic markers of neuroendocrine pathology[24].Several studies have shown that microvascular density is 10 to 30 times greater in NETs than in other carcinomas[25].
TUMOR ANGIOGENESIS
As previously mentioned, the most important tumor-induced angiogenesis mediator is VEGF and its receptors[9](Table 1).Six subtypes of VEGF are recognized: VEGF-A; VEGF-B; VEGF-C; VEGF-D; VEGF-E; and placental growth factor[26].VEGF-C and VEGF-D take part in lymphangiogenesis.VEGF-A plays a dominant role in the angiogenesis process and is simply referred to as VEGF[27].
VEGFgene transcription is regulated by hypoxia-inducible factor (HIF), which is a protein composed of a constant subunit (HIF-1β) and an oxygen-regulated subunit (HIF-1α or HIF-2α)[28].In response to hypoxia, the level of VEGF increases significantly in the extracellular space.High concentrations of VEGF determine the degradation of the basement membrane and the destabilization of the pericytes, the growth of endothelial cells, and the formation of new vessels[29].This process is highly involved in tumor progression and when small tumors receive their nourishment by passive diffusion[30].Those over 2 mm2undergo the formation of a hypoxic central core that stimulates the angiogenesis process[31].This phase is called the “angiogenic switch” and is the release of many mediators of angiogenesis by the tumor cells in response to the reduced oxygen supply[32].
There are different mechanisms by which neoplasms stimulate angiogenesis[33].The first and most important mechanism is germinal angiogenesis, which leads to the formation of new vessels from pre-existing capillaries and small venules.The endothelial cells undergo reactivation resulting in the formation of small shoots that grow and migrate into the adjacent connective tissue.Subsequently, an immature vessel is formed, stabilizing after the recruitment of pericytes and the reconstitution of the basement membrane.The new vessels are characterized by fenestrated endothelial cells, a discontinuous basement membrane, and rare pericytes.Consequently, the vascular network is permeable without efficient flow regulation and has an aberrant morphology with irregularly branched and tortuous vessels[34].
Another mechanism of tumor neovascularization is co-optation.In this case, the cancer cells grow along the normal vascular network.This mechanism is mainly observed in the brain, liver, and lung.It is particularly important in the early metastatic processes.Intussusception is the division of a pre-existing vessel into two new vessels and has been described in some aggressive tumors.Finally, in the vascular mimicry mechanism, a formation of vessels from the tumor cells themselves is observed.This process is seen in many aggressive tumors[35].
Pericytes are smooth muscle cells that stabilize the vessel walls and protect the normal vessels themselves from anticancer drugs, guaranteeing and promoting their target action.Hypoxia and angiogenic factors released by the tumor destabilize the pericytes and facilitate continued angiogenesis[8].The reduction in their number leads to an increase in permeability and consequently the interstitial fluid pressure[36].This leads to a further reduction in perfusion, the distribution of anticancer drugs, and acidosis[37].Interstitial fluid pressure can be considered a marker of response to anticancer therapy[38].
Hypoxia can promote chemoresistance by increasing the ATP-binding cassette efflux pumps.Hypoxic cells are less proliferative than their normoxic counterpart and are therefore less subject to the chemotherapeutic cytotoxic effect[39].Hypoxia also contributes to reducing the response to immunotherapy because it reduces immune activity[40].An increase in HIF1 levels prevents the activation of CD8+ T-helper lymphocytes, suppresses the cytotoxic effect of natural killer cells, and increases the expression of immunosuppressive mediators such as inducible nitric oxide synthase and interleukin (IL)-10 by dendritic cells.
Different therapeutic strategies have been developed in an attempt to make hypoxia an advantage.Drugs activated by an enzymatic reduction in a hypoxic environment with the production of cytotoxic compounds have been tested without a real confirmation in terms of clinical utility[41].Similarly, attempts were made to increase the oxygen transport capacity of the plasma using hyperbaric therapy[42].
In 1993, Kimet al[43] treated a mouse model of rhabdomyosarcoma, glioblastoma, and leiomyosarcoma with anti-VEGF monoclonal antibodies, obtaining tumor growth arrest.Given the ineffectiveness of these antibodiesin vitrothis pioneering study showed how blocking the action of angiogenesis mediators had a direct effect on tumor growth.However, the effect of these drugs was not constant[44].There are differences in antitumor responses based on dosage,duration of treatment, and tumor type.
Due to the tremendous vascularization that characterizes them, neuroendocrine lung tumors would most benefit from antiangiogenesis drugs.This observation refers to the architecture of normal endocrine glands that need a wellrepresented vascular network that allows continuous exchange between endocrine cells and the bloodstream including hormone secretion.
Another characteristic of NETs that would suggest an elective use of antiangiogenic therapy as the treatment of choice is their marked ability to synthesize and secrete elevated levels of VEGF-A[45].In this aspect, they mimic the endocrine cells with the secretion of peptide hormones[46].Pancreatic islet β cells show the secretion of elevated levels of VEGF-A,which appears to play a significant role in the development of the dense vascular network of normal endocrine tissues[47].VEGF-induced angiogenesis is also important for tumorigenesis and tumor progression of NETs.The angiogenic phenotype is necessary for the transition from hyperplasia[48], and it can be blocked pharmacologically[49].Even in this process, VEGF-A plays a decisive role[50].

Table 1 Proliferation, migration, and differentiation by several factors/inductors implicated in angiogenesis
The microvascular density of pancreatic NETs is higher in benign tumors than in malignant tumors and in this context is higher in low-grade than in high-grade malignant tumors.It is also characterized by a better prognosis.This observation is called the “neuroendocrine paradox.” To explain this phenomenon, it has been hypothesized that in pancreatic NETs the vascular density is a marker of differentiation rather than of aggressiveness[51].Like their normal counterpart, well-differentiated neuroendocrine cells do keep the ability to promote the formation of a dense vascular network.Conversely, the tumor angiogenesis mechanism of poorly differentiated neoplasms is secondary to hypoxia and aberrant genetic alterations.This does not signify the absence of angiogenic activity in well-differentiated NETs but that it is low per unit of time considered.
Little is known of the process of angiogenesis in NETs originating from organs other than the pancreas, and any available data are scarce and contradictory[52].As far as the lung is concerned, it appears to be similar to the pancreas,with the presence of high vascular density in well-differentiated NETs and low in high-grade NETs.However, all aspects are not yet completely clear, and further studies are needed, particularly in the area of high-grade and metastatic cancers where antiangiogenic therapies would find their main application.
Several antiangiogenic target drugs have been successfully assessed in metastatic NETs such as anti-VEGFA, anti-VEGFR, and tyrosine kinase inhibitors.However, other drugs already in use in the therapy of NETs have also shown an antiangiogenic action.Among these are the analogues of somatostatin and interferon alpha.Somatostatin analogues have shown antiangiogenic propertiesin vitroby inhibiting the proliferation of endothelial cells and the synthesis and secretion of VEGF.However, data on their usein vivoare controversial, probably due to their insufficient ability to compete with VEGF and other proangiogenic factors[53].The data in favor of the use of interferon alpha for the treatment of carcinoids seems more convincing.There is a significant reduction in intratumor microvascular density, but it is not associated with a reduction in circulating VEGF levels.
The development of resistance to antiangiogenic drugs is one of the major problems linked to their use, which is similar to other targeted therapies.This effect would explain the lack of long-term response and the so-called “angiogenic explosion” after their suspension.When anticancer drugs with antiangiogenic action are used at high dosages, they only have an acute antitumor effect that is not reflected long term.
Acute hypoxia due to massive and non-selective vascular destruction selects and facilitates only the most aggressive cancer cells, preventing immune surveillance, favoring metastases, and promoting resistance to anticancer treatments.Their use at low dosages as an adjuvant in chemotherapy regimens has instead shown efficacy thanks to the establishment of the so-called “vascular normalization” phenomenon[54].This consists of the selective destruction of only immature and aberrant vascularity while respecting the normal one.Vascular normalization also passes through the fortification of the vessel wall as a result of the recruitment of pericytes.Finally, antiangiogenic drugs also determine a tumor microenvironment[40] effect of normalization due to the reprogramming of many tumor processes that target blood vessels.
Several studies showed[55] that the biological basis of resistance is not found in the genetic mutations that occur in the target molecules but rather in the establishment of a secondary angiogenesis pathway.Malignant cells can simultaneously synthesize and secrete many proangiogenesis factors, among which angiopoietin-2 seems to be the one that plays the most important role.This alternative route was observed in the experimental models of NET[56] and could justify both the increase in serum levels of angiogenic cytokines during anti-VEGF/VEGFR therapy and the simultaneous and effective use of combined therapies that block multiple angiogenic routes.
The use of angiogenesis markers could be a promising way to monitor the efficacy of antiangiogenesis therapy,determine its optimal dosage, avoid related toxicity, and predict its response or resistance.Currently, microvascular density is the best-known tissue biomarker.However, many data from the literature[57] show that it is not predictive in response to antineoplastic therapy.Different approaches have yet to be explored using immunohistochemical, molecular,and serum methods.
OXIDATIVE STRESS
Eukaryotic cells obtain the energy needed from aerobic respiration in the mitochondria.Due to this metabolic process,several reactive species are produced.They are required for signal transduction, enzymatic activity, gene expression, and protein folding in the endoplasmic reticulum and during apoptosis.Commonly, they are harmless.However, about 5% of reactive species can be toxic if they reach high concentrations.
Biochemistry of oxidative stress
The sources of oxidative stress can be both internal and external to the cell.Peroxisomes and P450 complex enzymes,nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), xanthine oxidase, and NADPH complexes are all internal sources of oxidative stress.Almost all enzymes act within the mitochondria.Ultraviolet rays, chemicals (e.g.,environmental pollutants, smoking, and alcohol), and exercise are, conversely, external sources of oxidative stress.
Based on the main atom involved we can divide the reactive species into four groups: ROS; reactive nitrogen species(RNS); reactive sulfur species; and reactive chloride species[58].ROS and RNS are produced during the electron transport chain.ROS, which includes superoxide anion, hydrogen peroxide (H2O2), hydroxyl radical, singlet oxygen, and ozone, are the products of oxidative metabolism[59].Some ROS, such as peroxynitrite anion and ONOO-, can react with nitric oxide.Subsequently, nitric oxide is converted to a hydroxyl radical and a nitrite anion.
The balance between ROS and endogenous antioxidants determines the damage that cells can suffer.After the alteration of this balance, oxidative stress is generated with subsequent damage to DNA, RNA, lipids, and proteins[60].Reactive species cause DNA damage and malfunctions in the DNA repair mechanisms.The oxidation of DNA that takes place generates 8-hydroxy-2-deoxyguanosine, which is a product capable of causing mutations in DNA and increasing cellular aging and carcinogenesis[61].
Polyunsaturated lipids are abundant in the cell membrane and are also particularly susceptible to oxidation by reactive species.By peroxidation reactions, they release lipids and increase the permeability of the cell membrane, which can lead to cell death[62].However, proteins are the main target of the reactive species.The carbonyl (aldehydes and ketones) and thiol groups (–SH) can be converted into reactive sulfur radicals[63].Therefore, there is an alteration in the structure of the protein that leads to changes or loss of function.
The cell has three groups of defense mechanisms: endogenous antioxidants; natural antioxidants; and synthetic antioxidants[64].The following are endogenous antioxidants: glutathione; alpha-lipoic acid; coenzyme Q; ferritin; uric acid;bilirubin; metallothionein; l-carnitine; melatonin; superoxide dismutase; catalase; glutathione peroxidase; thioredoxin;and peroxiredoxin (PRX).PRX is a group of ubiquitous antioxidant enzymes (PRX I-VI).They can modulate the H2O2levels and transduce intracellular signaling.PRX III eliminates up to 90% of H2O2, and PRX V is even more effective against peroxynitrite.
The diet is a source of natural antioxidants such as ascorbic acid (vitamin C), tocopherol (vitamin E), carotene (vitamin A), lipoic acid, uric acid, glutathione, and polyphenolic metabolites.Finally, synthetic antioxidants include N-acetyl cysteine, thyroid hormones, pyruvate, selenium, butylated hydroxytoluene, butylated hydroxyanisole, and propyl gallate[65].
Clinical importance of oxidative stress
Several human diseases, such as neurodegenerative diseases (Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis), inflammatory diseases (arthritis), cardiovascular disease (atherosclerosis), allergies, immune system dysfunction, diabetes, aging, and cancer[66] are attributable to oxidative stress.During the acute inflammatory response,the chemical mediators released, such as ROS, also affect normal cells.In the case of a chronic inflammatory process,extremely high levels of ROS saturate the antioxidant mechanisms of the cell affecting the surrounding cells.
Oxidative stress in neoplasms
ROS are responsible for some cellular mechanisms implicated in tumor development and progression, including: (1) Cell proliferation (e.g., activation of regulated extracellular kinase 1/2 and ligand-independent kinase receptor tyrosine kinase); (2) Apoptosis inhibition; (3) Tissue infiltration and metastasis (metalloproteinase secretion in the matrix extracellular, Met overexpression, and Rho-Rac interaction); and (4) Angiogenesis (release of VEGF and angiopoietin).
Several biochemical pathways are affected by oxidative stress (from epidermal growth factor receptor to mechanistic target of rapamycin) involving key signaling proteins, such as Nrf2, Keap1, Ras, Raf, MAPK, ERK1/2, MEK, p38, JNK, cmyc, p53, and PKC[67-69].p38 acts as a key sensor of oxidative stress and is essential in the control of neoplastic development[70].Unlike other MAPKs, p38 suppresses tumorigenesis by blocking proliferation and promoting apoptosis(Table 2).
Genetics of oxidative stress in neoplasms
A key role in the neoplastic transformation is played by genetic factors.A high level of ROS is associated with the increased metabolism observed in tumor cells; however, oxidative stress is less harmful to cancer cells than it is to normal cells.Cancer cells can adapt to the new conditions and proliferate, creating a new redox balance.This ability of cancer cells allows them to have a greater resistance to oxidation and oxidative stress than normal cells.It follows that the neoplastic cells can increase their metabolic rate and proliferation and avoid the damage caused by free radicals.However, this adaptive response alone cannot explain the high metabolic rate of tumors[71].
Genetic factors implicated in tumorigenesis may also directly or indirectly modulate ROS levels.The physiologic antioxidant activity is mainly regulated by the Nrf2 transcription factor in addition to specific antioxidant enzymes, such as superoxide dismutase, catalase, glutathione peroxidase, thioredoxin, and PRX.Nrf2 modulates the expression of many genes, including not only those that code for antioxidant enzymes but also genes that control immune and inflammatory responses, carcinogenesis, and metastasis[72].ROS levels are controlled by Nrf2 and its repressor protein (Keap1).Furthermore, experimental data show that when treated with oxidation-inducing drugs Nrf2-free mice develop more severe intestinal inflammation than controls, suggesting a function for Nrf2 in preventing inflammation and carcinogenesis[73].
While Nrf2 was initially thought to be able to regulate oxidative stress by modulating the production of antioxidant enzyme antioxidant response element, subsequently kinase-dependent mechanisms have been described, such as MAPK,PI3K, and other alternative pathways for activation of Nrf2[74,75].Somatic mutations that disrupt the Nrf2-Keap1 interaction have been identified in patients with non-small cell lung cancer[76] and esophageal cancer[77].In breast cancer, the breast cancer tumor suppressor gene 1 (BRCA1) is mutated in 40%-50% of hereditary breast cancers, while it is absent or at a low level in 30%-40% of sporadic cases[78].BRCA1 is responsible for DNA repair and can regulate Nrf2 andNFκB[79,80].Nrf2 induces enzymes such as glutathione S-transferase, glutathione peroxidase, and oxidoreductase, which exert a protective action against ROS.In breast cancer cells theBRCA1gene reduces RNS damage to cells and helps them cope with oxidative stress.Redox factor 1/AP endonuclease 1 also participates in the reduction of ROS generation[81].

Table 2 Molecular target of oxidative stress to promote tumor progression
The Ras pathway (Ha-, N- and Ki-ras) is very important for regulating oxidative stress in cancer[82].Ras activating point mutations are present in tumor cells (approximately 30% of tumors), resulting in a constitutively active protein.These mutations lead to an increase in ROS levels, which induces neoplastic transformation[83].TheRasVal12 mutant activates the NOX4-p22phox NADPH oxidase system, which produces H2O2.Consequently, the response toRasVal12-induced DNA damage is impaired by the inhibition of NADPH oxidase.NADPH oxidase, NOX4, can be considered a critical mediator ofRasVal12-induced oncogenic DNA damage[84].
If theRasoncogene is overexpressed, cells show an increase in mitochondrial mass and an accumulation of ROS.Among these, the ROS generated by the respiratory chain in the mitochondria and the NOX enzymes in the cytoplasm are particularly important.NOX proteins are oncogenic proteins, and mitochondrial dysfunction is associated with tumorigenesis[85].
Mitochondrial dysfunction leads to DNA damage, decreased ATP levels, and activation of AMPK.The presence of theK-rasVal12 mutant in normal epithelial cells leads to increased peroxide levels and increased DNA damage.Peroxides can be generated by the COX-2 enzyme due to their correlation with K-ras[86].Consequently, the COX-2 enzyme is also involved in many human cancers.Both peroxide production and DNA damage are reduced by pretreatment with the COX-2 antagonist SC58125.Therefore, several proteins including COX-2 and the transcription factor HIF-1α, which is activated in response to low oxygen concentrations, can influence the oncogenic activity of mutant K-rasVal12.
Overexpression of oncogenic proteins [Raf, reverse transcriptase of Mos, MEK, Myc, cyclin E and human telomerase reverse transcriptase (hTERT)] and inhibiting oncosuppressor genes (p53, p21CIP1, PTEN) can cause aging by increasing ROS levels.PTEN deficiency and Ras/MAPK activation could promote metastasis and EMT from prostate precursor cells[87].Even in glioblastoma cells, PTEN deficiency, associated with high levels of Akt and ROS, leads to senescence.There is evidence that suggests thehTERToncogene acts by modulating the redox system[88].hTERT is localized in mitochondria, and its activity could influence the redox balance through the recruitment of the same mitochondria.Finally,hTERT inhibitors can induce mitochondrial-dependent apoptosis in target cells[89].
Many other genes are involved in regulating energy metabolism in cancer.p53, for instance, is one of the best-known tumor suppressors, and it is implicated in cellular energy balance in the mitochondria between glycolysis and the respiratory chain.Homologous cytochrome oxidase 2 is an important enzyme that mediates this effect, and its activity is very important for the regulation of the COX complex.Reduced homologous cytochrome oxidase 2 synthesis can cause low respiration and a high rate of glycolysis[90].
Sirtuins are a group of proteins involved in many cellular processes (aging, stress response,etc).Sirtuins are deacetylase enzymes regulated by NAD (positive activity) and NADH (negative activity).Sirt3 is the most studied of the three mitochondrial sirtuins and is known to act as a tumor suppressor.It is for this reason that it has been linked to longevity in humans.Kimet al[91] showed that in Sirt3 (-/-) murine embryonic fibroblasts, increased glycolysis,decreased oxidative phosphorylation, and increased ROS can be observed.Furthermore, the loss of Sirt3 increases cell tumorigenesis[92].This process is accompanied by the activation of the HIF-1α target gene under hypoxic conditions.
NEUROENDOCRINE LUNG TUMORS
Bronchopulmonary neuroendocrine neoplasms represent a group of rare neoplasms (accounting for almost 20% of all lung neoplasms)[93] arising from the proliferation of cells with both endocrine and nervous phenotypic characteristics that together form the diffuse neuroendocrine system[94].
Based on their morphological, structural, immunohistochemical, and ultrastructural characteristics, they can be divided into four groups according to the 5thedition of the World Health Organization classification on thoracic tumors[95]: typical carcinoid (TC); atypical carcinoid (AC); large cell neuroendocrine (LCNEC); and small cell carcinoma (SCLC).TC and AC are considered well-differentiated NETs, while LCNEC and SCLC are considered poorly differentiated tumors.TC and AC are low (corresponding to G1 NET) and intermediate (corresponding to G2 NET) grades,respectively, whereas LCNEC and SCLC are high grades (traditionally graded as G3 tumors).Although these four subgroups of neuroendocrine neoplasms may represent a continuum in the neuroendocrine differentiation spectrum,histological, immunohistochemical, and molecular studies have demonstrated that pulmonary carcinoids are different from poorly differentiated neuroendocrine carcinomas[96].
The first description of a bronchopulmonary carcinoid dates back to 1831 when Laennec[97], in his treatise on mediated auscultation of the lungs and heart, reported the case of a posthumous endobronchial mass.The clinical presentation can occur with cough, hemoptysis, and recurrent pneumonia (due to the functional exclusion of a bronchus by a growing mass) even if in most cases their clinical course is indolent[93].
The diagnosis is based on imaging methods, such as computed tomography and magnetic resonance imaging,bronchoscopy, bronchial biopsy or fine-needle aspiration biopsy, mediastinoscopy (in selected cases), scintigraphy with 111 In-pentetreotide (octreoscan), and functional studies such as the evaluation of the tumor secretion pattern.Although less than 5% of patients with bronchopulmonary carcinoids have symptoms such as carcinoid syndrome, Cushing’s disease, acromegaly, or syndrome of inappropriate antidiuretic hormone secretion, it is possible to detect secretion of amines, peptides, or hormones (endocrine, autocrine, or paracrine)[93].
However, the NETs most striking phenotypical characteristic is the massive vascularization[52] due to their marked ability to synthesize and secrete high levels of VEGF[45].The experimental data available refer especially to the pancreatic NETs where the presence of high vascular density in NETs and low vascular density in neuroendocrine carcinoma is observed.The precise situation and the angiogenesis mechanism is not completely clear in neuroendocrine lung tumors.This review could provide a starting point for further future studies.
Experimental evidence has shown that the ROS released by the tumor due to metabolic stress are associated with different outcomes depending on their level[31].Evidence shows that high levels of ROS directly lead cancer cells to cell death whereas low to medium ROS levels increase neoplastic progression, metabolism alteration, cell migration, EMT,and metastasis[98,99].ROS also stimulate acute inflammation that becomes chronic when associated with prolonged ROS production[100].NFκB and TGF-β are implicated in the relationships between chronic inflammation and carcinogenesis[101].ROS are also responsible for p38 MAPK activation and TGF-β1-mediated EMT in many tumors[14].Mitochondria are very important in determining neoplastic degeneration due to their production of endogenous ROS that subvert the metabolic process and oxidative phosphorylation[102].
Oxidative stress induces the production of ROS-dependent cytokines such as TGF-β, IL-6, IL-13, and VEGFA.A change to the mitochondrial redox and consequently the acid-base balance of the tumor microenvironment could represent a therapeutic strategy to improve the cellular function of T lymphocytes during immunotherapy treatment[103].
CONCLUSION
The use of angiogenesis and oxidative stress markers could be useful for evaluating the efficacy of antineoplastic drugs,establishing the optimal dosage, escaping from the related toxicity, and predicting its response or resistance.
FOOTNOTES
Author contributions:Andriolo LG and Cammisotto V designed the research study; Andriolo LG and Di Rienzo G performed the research; Andriolo LG, Cammisotto V, Spagnoli A, and Alunni Fegatelli D analyzed the data and wrote the manuscript; All authors read and approved the final manuscript.
Conflict-of-interest statement:All the authors declare that they have no conflicts of interest.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial.See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:Italy
ORCID number:Luigi Gaetano Andriolo 0000-0002-1094-5118; Vittoria Cammisotto 0000-0003-1966-5945; Alessandra Spagnoli 0000-0002-7772-3130; Danilo Alunni Fegatelli 0000-0001-9726-8617; Michele Chicone 0000-0002-1708-4023; Pasquale Pignatelli 0000-0002-2265-7455.
S-Editor:Liu JH
L-Editor:Filipodia
P-Editor:Yu HG
杂志排行

World Journal of Meta-Analysis的其它文章
- Evidence relating cigarettes, cigars and pipes to cardiovascular disease and stroke: Meta-analysis of recent data from three regions
- Endoscopic vs radiologic gastrostomy for enteral feeding: A systematic review and meta-analysis
- History, origin, transmission, genome structure, replication, epidemiology, pathogenesis, clinical features,diagnosis, and treatment of COVID-19: A review