环己胺合成研究进展
2023-11-15张美华钟齐锋陈梦婷刘迎新
张美华,钟齐锋,陈梦婷,刘迎新
(浙江工业大学 药学院,浙江 杭州 310014)
伯胺作为一种通用的平台分子,已被广泛应用于化学工业和生命科学[1]。作为伯胺类化合物的代表物质之一,环己胺在医药、农药、食品添加剂等领域应用广泛[2]。环己胺是合成多种药物的关键中间体[3-5];环己胺的磺酸盐(甜蜜素),作为一种低成本甜味剂,用于食品、饮料等行业[6];环己胺可用于生产N-环己烷基-2-苯并噻唑次磺酰胺促进剂[7]。近年来,随着环己胺下游产品的不断开发,环己胺需求量正逐步增加。工业上,环己胺通常经两步法生产:1)硝基苯还原为苯胺;2)苯胺氢化为环己胺。然而,该合成方法条件苛刻且步骤复杂,缺乏可持续性和绿色性。其他常规合成方法(如烯烃氢胺化、卤代环己烷催化氨解和醇借氢胺化等)的起始材料多数来源于不可再生的化石资源、原子经济性低且易产生废盐。近年来,随着绿色化学的发展,以可再生木质素衍生的平台分子高效合成环己胺的研究受到了广泛的关注。与传统途径相比,绿色木质素基合成路线不仅具有较少的合成步骤,而且原子经济性高,能够显著提高成本竞争力、降低能耗。
本文根据所用原料的不同,综述了近年来较为常见的通过催化官能团转换反应合成环己胺的方法,包括苯胺催化氢化法、硝基苯催化氢化法、环己醇还原胺化法、环己酮还原胺化法、酚类还原胺化法等。介绍了各个方法所使用的催化体系、反应机理并比较了不同方法的优缺点。最后对环己胺的发展前景进行展望。
1 环己胺的主要合成方法
1.1 苯胺催化氢化法
苯胺催化氢化法是目前合成环己胺最常用的方法之一。一般来说,苯胺催化氢化合成环己胺具有100%的原子利用率。反应机理如图1 所示,除环己胺外,产物中还可能存在二环己胺和环己醇等副产物[8]。

图1 苯胺催化氢化合成环己胺的反应机理Fig.1 Reaction mechanism of catalytic hydrogenation of aniline to cyclohexylamine.
由于苯环的高稳定性,苯胺氢化转化使用的催化剂主要是贵金属基催化剂。Chatterjee 等[9]在超临界CO2中,使用贵金属(Pt,Pd,Rh)催化剂将苯胺氢化转化为环己胺。在80 ℃、8 MPa CO2、4 MPa H2下反应6 h,在5%(w)Rh/Al2O3催化剂上苯胺转化率为95%,环己胺选择性为93%。在超临界CO2中的高环己胺选择性和高转化频率(223.3 h-1)表明了CO2扩增底物的重要性。实验结果表明,CO2与环己胺相互作用形成固体氨基甲酸,从而提高了环己胺的选择性。但由于固体氨基甲酸的沉积导致相分离,苯胺的转化率显著降低。该催化体系以CO2为加氢“替代”媒介,避免了传统氢化过程中氨和/或有机溶剂的使用。然而高达12 MPa 的反应压力,无疑会增加安全问题。Cao 等[10]制备了具有Lewis 酸碱对的Ru0-Ruδ+/CeO2催化剂。在100 ℃、2 MPa H2下反应200 min,苯胺转化率为95%,环己胺选择性大于99%。环己胺的高选择性主要归功于Lewis 酸碱位点在CeO2上的共存[11]。酸性载体上吸附的相邻胺分子可能会发生缩合反应[12]。而在CeO2上,具有Lewis 酸性位点的Ce 原子被表面或内部的碱性氧位点所包围(甚至被氧空位所增强),形成“酸性位点分离”,有效地抑制了胺的缩合反应。该催化剂对不同官能团取代的芳胺和杂芳烃的氢化表现出较高适用性,相应的胺收率为80%~99%。非贵金属Ni 基催化剂[13]和Co 基催化剂[14]在苯胺的氢化中应用广泛。但这些催化剂通常表现出较差的选择性和低稳定性。Murugesan 等[15]使用廉价的焦甲酸(PMA)和哌嗪(PZ)连接剂制备了Co 基配位聚合物并将其组装在SiO2上,得到了特定的Co纳米催化剂(Co-PMA- @SiO2-800)并用于苯胺的催化氢化反应。在135 ℃、5 MPa H2下反应24 h,环己胺收率为90%。该催化剂对取代和功能化芳烃和酚及多环芳烃的氢化反应具有普遍适用性。目前,虽然已开发了一系列的催化剂用于苯胺的催化氢化,但是苯环的稳定性使得大多数催化体系的反应条件较为苛刻,需要高温、高压、较长的反应时间等。因此,开发在温和条件下实现苯胺加氢制环己胺的非贵金属催化体系将是未来研究的主要任务。
1.2 硝基苯催化氢化法
硝基苯一锅法催化氢化生成环己胺的过程结合多种化学转化,但无需进行苯胺的分离和纯化,经济简便。硝基基团强烈的吸电子作用导致硝基苯的芳环缺电子[16]。因此,硝基苯的芳环与金属的配位性较弱[17]。相比之下,硝基具有很强的配位性[18]。所以硝基苯中的硝基具有很强的先氢化倾向(图2,路径A/B);当然,芳环也可先被氢化(图2,路径C/D)[19-20]。相比于苯胺的直接氢化,硝基苯氢化的反应路径更为复杂,涉及多条反应路径,可能生成亚硝基苯、苯基羟胺和氢化偶氮苯等多种副产物[21]。
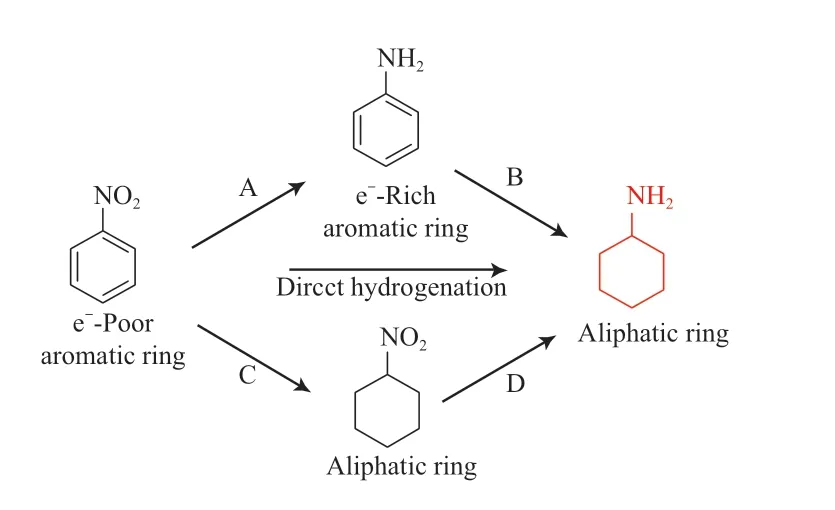
图2 硝基苯催化氢化合成环己胺的反应路径Fig.2 Reaction pathway of catalytic hydrogenation of nitrobenzene to cyclohexylamine.
Tomkins 等[22]制备出碳纳米管(CNT)负载的催化剂Pu/CNT 和Pt/CNT,将两种催化剂以Pt/Ru摩尔比5∶95 混合,在140 ℃、10 MPa H2下反应0.4 h,硝基苯转化率为100%,环己胺选择性高达92%。使用催化剂混合物而不是双金属催化剂,可避免相分离和其中一种金属迁移到双金属粒子的表面。许多研究发现,特定的添加剂可抑制硝基苯催化氢化合成环己胺反应过程中副产物的形成,但机理尚不清楚[19,23]。Li 等[21]系统研究了添加剂对Ru/活性炭催化剂催化硝基苯加氢反应的影响。通过加入碱金属氢氧化物、亚硝酸盐和硝酸盐,90 ℃、6 MPa H2条件下反应50 min,环己胺的选择性从85.4%提高到100%。实验结果表明,不同添加剂对副反应的抑制实质上是氢氧根离子的作用。理论计算进一步表明,碱金属氢氧化物通过降低环己胺的吸附能来降低烯胺加氢的能垒,释放出更多的活性位点,有利于芳基的加氢。环己胺更容易从活性位点解吸,从而阻止它与亚胺的反应。Chaudhari等[24]报道Pd0.5Ru0.5-聚乙烯吡咯烷酮(PVP)催化剂在温和反应条件下催化硝基苯加氢一锅法合成环己胺。在100 ℃、2 MPa H2下反应6 h,硝基苯转化率为99%,环己胺收率为92%。催化剂重复使用3 次后,催化活性无明显下降。在优化条件下,Pd0.5Ru0.5-PVP 对多种官能团取代的硝基苯具有较高的氢化活性,相应的胺收率为60%~75%。C3N4是一种特殊的掺氮碳材料,氮物种不仅提供了金属配位环境,而且含有丰富的碱性位点可抑制副反应[25]。Wu 等[26]将Ru,Pd 纳米颗粒锚定在C3N4上,制备了具有Ru 和Pd 双活性位点和碱性N 位点的1.5%(w)Ru-1.5%(w)Pd/C3N4-空气催化剂并用于硝基芳烃在温和条件下连续加氢生成环己胺。在80 ℃、3 MPa H2下反应3 h,硝基苯转化率为100%,环己胺选择性为96.8%。实验结果表明,Pd 是硝基加氢的主要活性金属,而Ru 是苯环加氢的主要活性金属。同时,相应的非优势金属增强了H2的活化和解离,从而显著提高了催化活性。催化剂循环实验表明,在8 次循环中,硝基苯保持100%的转化率,环己胺的选择性从最初的95.2%下降至87.3%。使用后催化剂的TEM 照片显示,这种轻微的催化剂活性下降可归因于催化剂边缘的小部分金属纳米粒子团聚。Ru-Pd 催化剂的优异性能可归功于Ru-Nx和Pd-Nx在纳米颗粒上的高度分散和Ru-Pd 与C3N4-空气之间强烈的金属-载体相互作用,抑制了活性金属的团聚,从而维持催化活性。该催化体系完全避免了碱性添加剂的使用,对环境友好。Lu 等[27]在掺杂0.3%(w)Rh的10%(w)Ni/生物碳(CSC)催化剂上采用硝基苯一锅法高选择性液相加氢生成环己胺,在140 ℃、3.5 MPa H2下,添加40 mg 氢氧化锂,反应6 h,硝基苯转化率为100%,环己胺选择性为91.6%。0.3%(w)Rh-10%(w)Ni/CSC 催化剂重复使用6次后,催化活性无明显下降。此外,该催化剂对一系列取代硝基苯的氢化反应具有催化活性,相应的胺收率为72.9%~94.4%。环己胺的高选择性归因于加入的碱性添加剂(氢氧化锂)中和了碳载体表面的酸性位点,抑制胺类中间体在载体上的强吸附。Lu 等[28]采用微波加热法制备了生物碳负载的Ni 基催化剂(10%(w)Ni/CSC),用于硝基苯选择性加氢生成环己胺。在200 ℃、2.0 MPa H2下反应4 h,硝基苯转化率为100%,环己胺收率为96.7%。催化剂可重复使用6 次,且无明显活性损失。催化剂对多种官能团取代的硝基苯具有较高的氢化活性,相应的胺收率为78.3%~96.7%。该催化体系完全避免了Rh 等贵金属的使用和碱性添加剂的加入,但200 ℃反应温度,无疑给工业化生产带来了更大的挑战。
综上所述,目前关于硝基苯催化氢化的研究主要集中在贵金属Ru 基和Ni 基催化剂。金属Ru 和Ni 的作用是有限的,通常需要添加第二金属或碱性添加剂来提高硝基苯的转化率和环己胺的选择性。然而,双金属的使用增加了反应成本,添加剂在提高环己胺的选择性的同时也抑制了催化剂的活性。非贵金属Ni 基催化剂通常需要与较高的反应温度(200 ℃)相结合。因此开发高活性和高选择性的且无添加剂的新型非贵金属催化体系将是未来研究的主要目标。
1.3 环己酮还原胺化法
羰基化合物的还原胺化具有操作简单、反应条件温和和底物来源广泛等优点[29-30]。因此,在制药工业中至少有四分之一的C—N 键通过还原胺化反应形成[31-32]。环己酮的还原胺化反应机理如图3 所示[33]:1)氨对羰基亲核攻击,形成亚胺中间体,2)亚胺中间体进一步氢化生成环己胺。H2是环己酮还原胺化中最常用的氢源,因为H2的存在可减少金属氮化物的形成,并帮助维持催化剂的活性[34-35]。

图3 环己酮还原胺化合成环己胺的反应机理[33]Fig.3 Reaction mechanism of reductive amination of cyclohexanone to cyclohexylamine[33].
均相催化体系具有明显的缺陷性,如催化剂回收困难、使用添加剂和配体及苛刻的反应条件(4.0 ~6.5 MPa H2和120 ~135 ℃)[36-38]。对于非均相催化剂,目前已经研究了Ru,Co,Cu,Ni基催化剂等催化环己酮的还原胺化反应。Xie 等[29]以天然存在的植物酸为可再生前体,制备了磷酸钛(TiP)负载的Ru 纳米催化剂(Ru/TiP-100),并用于常温下酮的还原胺化反应。在30 ℃、1.4 MPa H2、0.6 MPa NH3下反应6 h,环己酮转化率为100%,环己胺收率为97%。此外,底物拓展实验表明超过20 多种羰基化合物均可在优化条件下转化为相应的胺,收率为83%~98%。催化剂的高活性归因于Ru 物种上适宜的电子密度可平衡加氢过程中H2的活化和中间体(亚胺和Schiff 碱)的解吸。同时,Ru/TiP-100 相对较高的酸度可促进原位生成的亚胺和Schiff 碱中的C=N 基团转化为所需的伯胺。Qin 等[39]在固定床反应器中研究了Cu-Cr-La/γ-Al2O3催化剂对环己酮与氨的还原胺化反应的影响。在180 ℃、3 MPa H2下,环己酮的转化率为99.0%,环己胺的选择性达到83.9%。实验结果表明,在固定床反应器中加入氨可明显提高环己胺的选择性。分子筛的加入可通过去除水来极大地促进环己胺的形成。与简单的加氢化反应不同,酮的还原胺化要求催化剂能够在过量的氨或胺的存在下发生,这些氨或胺通常强烈吸附在金属催化剂上,导致催化剂中毒[40]。因此,催化剂的稳定性是催化剂研究的重点。Zhang 等[41]利用简单的共沉淀法制备了Ni1Al 纳米催化剂,并将其应用于羰基化合物的还原胺化。以环己酮为底物,在100℃、2 MPa H2下反应1 h,环己胺收率为97%。Ni1Al 催化剂由于良好的抗烧结和抗氧化性能,在连续流反应器中也表现出优异的稳定性,在200 h 内保持90%以上的胺收率。TEM,H2-TPD,NH3-TPD 等表征结果显示,Ni0纳米颗粒的良好分散性与表面丰富的酸性位点的协同作用是Ni1Al 催化剂具有高性能的主要原因。此外,在80 ~100℃和1 ~2 MPa H2的温和条件下,该催化剂对多种醛和酮的还原胺化具有较高的催化活性,目标胺收率为91%~99%。Zheng 等[42]通过金属有机框架ZIF-67 制备了Co@C-N(800)催化剂。在35 ℃、1.4 MPa H2、0.6 MPa NH3条件下反应6 h,环己胺收率为99%。该催化剂可至少重复使用5 次而没有明显的活性损失。此外,Co@C-N(800)催化剂具有良好的磁性,可很容易地用磁铁快速分离。除了使用非贵金属催化剂和较低的反应温度外,该催化剂的另一个独特优势是适用30 多种不同取代基的羰基化合物在低温下转化为相应的胺。表征结果显示,该催化剂由石墨烯封装的Co 纳米颗粒和原子分散的Co-Nx位点组成。Co 纳米颗粒促进了加氢反应,而Co-Nx位点作为酸性位点激活中间体(Schiff 碱)的进一步转化。
综上所述,环己酮的还原胺化法具有工艺绿色、原子经济性高的优点。尤其是非均相催化体系,由于反应条件温和、收率高且催化剂易于回收利用,从而具有较高的应用价值。然而,环己酮还原胺化过程中过量的氨对催化剂的活性产生了极大的抑制作用。单原子催化由于具有明确的活性位点结构和最大化的金属原子利用而成为非均相催化的前沿[43]。未来研究可尝试制备非贵金属单原子催化剂,在提高催化剂稳定性的同时有望得到高环己胺收率。
1.4 环己醇还原胺化法
环己醇的催化胺化遵循借氢法的基本步骤[44]:1)环己醇脱氢生成环己酮;2)环己酮与氨加成-消除生成环己亚胺;3)环己亚胺被从醇中提取的氢迅速还原生成环己胺(图4)。由于形成的环己胺具有更强的亲核性,反应过程中易生成二环己胺[45]。如果反应温度过高(高于 200 ℃)环己胺还可能进一步脱氢生成苯胺[46]。此外,由于醇脱氢的速率较慢,醇胺化过程通常反应时间较长(大于24 h)[47-48]。
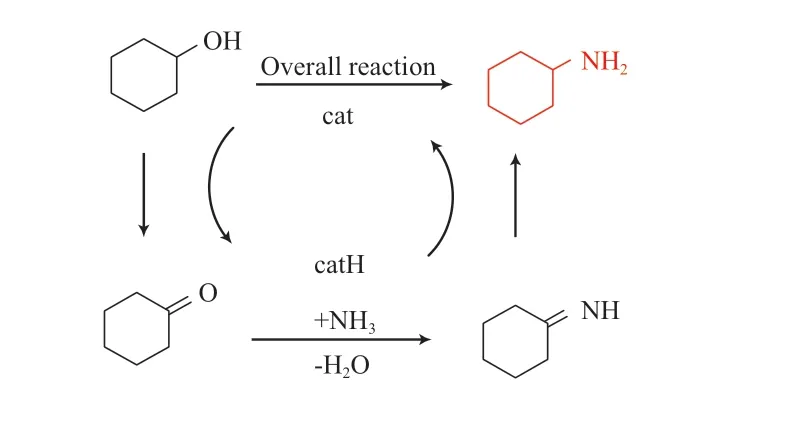
图4 环己醇还原胺化合成环己胺的反应机理Fig.4 Reaction mechanism of reductive amination of cyclohexanol to cyclohexylamine.
均相催化体系已被报道用于醇的还原胺化,主要包括Co 配合物[49]、Ru 基配合物[50]、Ir 基配合物[51]。然而,此类催化剂对环己胺的选择性不高。此外,多数催化剂或配体对空气或水分不稳定,导致催化剂回收困难。基于这些原因,醇胺化的非均相催化体系得到了广泛的研究。其中,Ni 基催化剂具有较大的可用性和低经济成本,且对环己胺具有较高的选择性,因此具有广泛的应用前景。Shimizu 等[52]研究表明,Ni/Al2O3是醇胺化成伯胺的首选催化剂。在无外加氢源的条件下,160 ℃下反应28 h,环己醇转化率为96%,环己胺收率可达到96%。环己胺选择性高达100%,但有毒且高沸点(144 ℃)的邻二甲苯溶剂对环境并不友好。此外,由于羟基的存在,醇在邻二甲苯中的溶解度受到限制。Qi 等[53]研究了Raney Ni,Ni/Al2O3,Ni/C 三种催化剂在水中和低沸点溶剂中将环己醇转化为环己胺。研究发现碱、氢、溶剂和载体都会影响催化剂的活性。在水中,三种催化剂在高温(160 ~180 ℃)及碱和氢存在下,均得到了85%以上的环己醇转化率和90%以上的环己胺选择性。在四氢呋喃和环己烷中,Ni/Al2O3的催化活性高于Ni/C。而在氨水中,Ni/C 的催化活性高于Ni/Al2O3。由此可见,溶剂对催化剂的催化活性有极大的影响。Tong 等[54]制备出1%(w)Pt-1%(w)Co/CeO2催化剂,用于醇的借氢胺化。在180 ℃下反应8 h,环己醇转化率大于99%,环己胺收率为73.1%。而在1%(w)Pt/CeO2催化剂上环己醇转化率大于99%,环己胺收率仅为32.2%。在优化条件下,1%(w)Pt-1%(w)Co/CeO2催化剂对伯醇、仲醇和苯甲醇的催化活性一般,相应伯胺收率为53.0%~57.0%。Wei 等[55]制备了水滑石衍生的NiCu/MgAlO 合金催化剂,并用于在固定床反应器中催化环己醇还原胺化合成环己胺,在170 ℃下运行300 h 后,仍保持98%的环己醇转化率和95%的环己胺收率。在140 ~210 ℃范围内,NiCu/MgAlO 对各种一系列环脂肪醇(酮)和链脂肪醇或酮的胺化反应具有较高活性,目标胺收率为45.6%~99.8%。模拟计算结果表明,在NiCu 合金催化剂中,Ni 是环己醇的脱氢活性位点,Cu 是环己亚胺的加氢活性位点。NiCu/MgAlO 催化剂的超高活性和稳定性归因于具有高分散和均匀粒径(约4 nm)的Ni-Cu 合金纳米颗粒。NiCu/MgAlO的高稳定性,使其具有广阔的工业应用潜力。
综上所述,环己醇的还原胺化反应具有底物来源广泛、原子经济性高的优点。虽然基于过渡金属的多用途非均相催化剂已经被开发出来,但它们只在特定的底物范围内才有效,且高温(高于140 ℃)是必要的。水是最绿色的溶剂,但是目前为止非贵金属在水中催化醇胺化的活性均不理想。因此,开发具有高稳定性且对于醇底物具有较大适用范围的催化剂,将是该方法未来的主要研究任务。
1.5 酚类还原胺化法
木质素是一种天然酚类聚合物,是自然界中唯一大量可用且可再生的生物质资源。苯酚、愈创木酚、二苯醚是木质素常见的模型化合物。由于木质素易于还原,被认为是合成环己胺的潜在结构单元。同时,亲电的C=O 在交叉偶联领域的突破性进展,促进了酚与不同亲核试剂直接偶联的可能[56]。因此,木质素衍生的酚类化合物还原胺化合成环己胺是一条有潜力的绿色可持续工艺路线。而且环己胺具有较高的经济价值,也是从木质素中获得净利润最高的化学品[57]。由于木质素模型化合物的固有性质,酚类化合物一锅法直接催化胺化通常需要较长的反应时间(12 ~24 h),且催化效率不理想,易产生多种副产物(图5)[58]。

图5 苯酚还原胺化合成环己胺的反应机理Fig.5 Reaction mechanism of reductive amination of phenol to cyclohexylamine.
目前,关于酚类化合物一锅法催化胺化的研究主要集中在Rh 基和Pd 基催化剂上。Tomkins等[59]以H2为氢源,探索了苯酚还原胺化的不同贵金属催化剂。在相同的反应条件下,Pd/C,Pt/C,Rh/C 催化剂对环己胺的选择性由小到大顺序为Pd/C 图6 Raney Ni 催化剂上愈创木酚还原胺化合成环己胺的反应机理Fig.6 Reaction mechanism of reductive amination of guaiacol to cyclohexylamine over Raney Ni catalyst. 综上所述,木质素模型化合物及其衍生物一锅法催化胺化研究取得了重大突破。但由于木质素模型化合物的结构特性[64],催化胺化发展仍旧缓慢。Ni基催化剂在苯酚的还原胺化显示出高催化活性,但催化剂稳定性一般。因此,开发一种低负载、高活性、高选择性、对底物和氮源耐受性强的非贵金属催化体系,是木质素基胺化合成环己胺的重要目标;有机溶剂(如甲苯、二甲苯)是酚类化合物还原胺化常用的溶剂,但这些溶剂往往是有毒的,如果大量使用,会造成严重的环境影响。使用水作为绿色溶剂不但可降低反应成本,还可大大降低有机溶剂造成的毒性和环境污染。鉴于某些胺化反应可在气相或无溶剂条件下有效进行,开发木质素底物的绿色溶剂或无溶剂胺化体系对可持续发展具有重要意义;H2和甲酸钠是酚类还原胺化法常用的氢源,但是氢分子的运输、储存和操纵可能存在严重的安全性问题。甲酸钠的使用会产生大量CO2。从安全和环保生产的角度来看,开发其他绿色氢源(如醇和水),可有助于提高生产的可持续性,并提高市场竞争力;环己胺具有较强的亲核性,很容易继续反应生成仲胺,因此一个具有比环己胺更强亲核性的氮源(如水合肼),对提高环己胺的选择性也非常重要。 硝基环己烷催化氢化法通常需要高温高压及贵金属催化剂(Au,Pd,Pt)[65-67]来确保反应顺利进行,且硝基环己烷原料不易得。氯代环己烷催化氨解极易发生过烷基化反应,导致产生伯胺、仲胺和叔胺及季铵盐的混合物[68],通常需要过量的氨抑制该副反应。此外,反应生成物中有HCl,对设备有腐蚀性。环己烯直接胺化法需要较为苛刻的反应条件(10.5 MPa H2、280 ℃),且环己胺收率非常低(4.3%)[69]。苯与羟胺一锅法合成,以强致癌性的苯为底物,因此并不常用[70]。上述方法经济性较低,并不适用于工业化生产。 苯胺催化氢化法、硝基苯催化氢化法、环己醇还原胺化法、环己酮还原胺化法、酚类还原胺化法和其他方法的环己胺合成工艺各有优劣。相较而言,尽管酚类还原胺化法面临反应产物复杂、反应条件苛刻的缺陷,但可再生的起始原料、较少的合成步骤和良好的环己胺收率,仍旧使其具有较大的工业化潜力。虽然环己胺合成研究已经取得了很大进展,但实现工业化应用仍然面临不小的挑战,在未来的研究中应该着重关注以下几个方面:1)苯胺和硝基苯的催化氢化法都具有较高的原子利用率,苯环的高稳定性使得苯胺和硝基苯的催化氢化主要集中于贵金属催化剂,但地球上贵金属储存有限,且价格昂贵。氢化过程中有毒的偶氮苯及其衍生物的形成仍然是一个很大的缺点。因此,需要开发一种具有高活性、高选择性和高稳定性的新型催化剂,尤其是非贵金属催化剂,从而提高环己胺的选择性。2)环己酮和环己醇的还原胺化法都具工艺绿色环境友好的特点,理论上都以产生水为副产物。尤其是醇的还原胺化可不依靠外加氢源,避免了对氢供体的依赖,具有更高的原子经济性。但是醇酮的还原胺化通常需要过量氨来提高环己胺的选择性,这无疑给催化剂的稳定性带来巨大的挑战。因此发展高稳定性的催化剂是这两种方法未来发展的主要目标。3)由于生物基分子来源于生物质衍生的碳水化合物,未来的研究重点可转向直接以生物基碳水化合物,甚至木质纤维素生物质,作为合成环己胺的起始材料。实现这一目标的关键是设计创新的集成过程,将碳水化合物高效转化为平台分子,并随后有效地促进生物基分子还原胺化为环己胺。
2 其他合成方法
3 结语
