盐酸羟考酮原料药中元素杂质控制方法研究*
2023-11-14田向斌王文丽张晓明
田向斌,柯 静,王文丽,张晓明
(甘肃省药品检验研究院,甘肃 兰州 730070)
盐酸羟考酮是一种强效镇痛药,是从生物碱蒂巴因中提取合成的阿片类药物,临床应用广泛[1-2]。盐酸羟酮缓释片主要作用于中枢神经,起效快,维持时间长,且不易产生不良反应[3-5]。2020 年版《中国药典(二部)》[6]中采用重金属检查法测定盐酸羟考酮中重金属限量。重金属限量检测在线性范围、准确性、灵敏度、专属性等方面均存在不足,多国药典中提出元素杂质控制的新标准和新方法[7-8]。人用药品注册技术要求国际协调会(ICH)Q3D 中将元素分为三类,其中第一类元素砷、镉、汞、铅有明显毒性,一般情况下在药物的生产中限制使用或不用,但会以杂质形式出现在原料中,且不易除掉[9]。本研究中采用微波消解法对样品进行前处理,采用光谱法[石墨炉原子吸收光谱(GF-AAS)法和原子荧光光谱法]和电感耦合等离子体质谱(ICP-MS)法对第一类元素及盐酸羟考酮原料药合成工艺中催化剂钯元素进行了分析,建立了盐酸羟考酮原料药中元素杂质的控制方法[10-15]。现报道如下。
1 仪器与试药
1.1 仪器
ICP - MS2030 型电感耦合等离子体质谱仪(日本Shimadzu公司);Agilent 240ZAA型石墨炉原子吸收分光光度计(美国Agilent公司);2100AFS型双通道原子荧光光度计(北京海光仪器有限公司);MiliQ7000 型超纯水系统(美国Millipore 公司);Anton Paar MultiwavePRO 型微波样品制备系统(奥地利Anton Paar 公司);ME204 型电子天平(瑞士Mettler-Toledo公司,精度为万分之一)。
1.2 试药
硝酸(批号为190304106,含量为68.0%~70.0%),过氧化氢(批号为190106103,含量为30.0~32.0%),均为金属- 氧化物- 半导体电路专用的特纯试剂,购于苏州晶瑞化学股份有限公司;盐酸(批号为20170406,含量为36.0%~38.0%),硼氢化钾(批号为20170915),硝酸镁(批号为20180909),均为优级纯,购于天津市科密欧化学试剂有限公司;氢氧化钾(优级纯),磷酸二氢铵(分析纯),硫脲(优级纯),抗坏血酸(优级纯),均购于国药集团化学试剂有限公司;盐酸羟考酮(批号分别为1708026-1,1708030-1,1708031-1,1708032-1,1708033-1,1810001),蒂巴因(批号分别为 201511011,201511012,201512013,201512014,201512015,201512016),均购于甘肃普瑞制药有限公司;ICP-MS Internal Std Mix(Agilent Technologies,质量浓度为100 μg/ mL);铅单元素标准溶液、砷单元素标准溶液(1 000 μg/mL)、汞单元素标准溶液、镉单元素标准溶液,质量浓度均为1 000 μg/mL,均购于中国计量科学研究院;钯单元素标准溶液(国家有色金属及电子材料分析测试中心,质量浓度为1 000 μg/mL)。
2 方法与结果
2.1 微波消解条件
条件见表1。

表1 微波消解条件Tab.1 Conditions of microwave digestion
2.2 工作条件
2.2.1 光谱法
详见表2。其中,镉元素基体改进剂为1%磷酸二氢铵+ 0.2%硝酸镁(共进10 μL)。砷元素和汞元素的负高压均为270 V,载气流速均为400 L/ min;采样时间为15 s。

表2 光谱法工作条件Tab.2 Working conditions of the spectrometry
2.2.2 ICP-MS 法
碰撞池模式:KED;高频功率:1.20 kW;采样深度:5.0 mm;等离子体气流速:8.0 L/ min;辅助气流速:1.10 L/min;载气流速:0.70 L/min。
2.3 溶液制备
2.3.1 光谱法
稀释液:取适量硝酸和盐酸溶液,用超纯水稀释成2%硝酸+0.5%盐酸的稀释液。
标准贮备液:精密吸取铅单元素标准溶液适量,用稀释液稀释成每1 mL 含铅20 ng的标准贮备液;精密吸取钯单元素标准溶液适量,用稀释液稀释成每1 mL 含钯100 ng 的标准贮备液;精密吸取镉单元素标准溶液适量,用稀释液稀释成每1 mL含镉5 ng的标准贮备液;分别精密吸取汞单元素标准溶液、砷单元素标准溶液适量,置同一容量瓶中,用稀释液稀释成每1 mL 含汞200 ng、砷4 000 ng的混合贮备液。
系列标准溶液:分别吸取2.3.1项下铅、钯、镉标准贮备液,以稀释液为标准空白溶液,由石墨炉原子吸收分光光度计自动稀释进行标准曲线测定。取2.3.1项下汞、砷元素混合贮备液,用稀释液稀释成每1 mL分别含汞0.1,0.2,0.4,0.8,1.6,3.2 ng,含砷2,4,8,16,32,48 ng的系列混合标准曲线溶液。
供试品溶液:取样品0.5 g,精密称定,置微波消解罐中,加入硝酸5 mL,过氧化氢溶液2 mL,置微波消解仪中,按消解程序将样品消解完全,放冷,取出,置控温电热板上,130 ℃加热使多余的酸挥发除尽,并持续浓缩至1~2 mL,放冷,转移至25 mL 容量瓶中,用稀释液稀释并定容。
空白溶液:同供试品溶液制备方法制备不加样品的试剂空白溶液。
2.3.2 ICP-MS 法
贮备液:分别精密量取铅、砷、镉、钯标准溶液各适量,用2.3.1项下稀释液制成每1 mL 含4 000 ng的混合贮备液;精密量取汞单元素标准溶液适量,用2.3.1 项下稀释液稀释成每1 mL含汞1 000 ng的标准贮备液。
系列混合标准溶液:分别取2.3.2项下混合贮备液和汞标准贮备液各适量,用2.3.1项下稀释液稀释成每1 mL 含铅、镉、砷、钯元素2,10,20,30,40,50 ng,含汞1,2,4,6,8,12 ng的系列混合标准溶液。
供试品溶液:同2.3.1项下。
空白溶液:同2.3.1项下。
2.4 方法学考察
线性关系考察:1)光谱法。取2.3.1项下系列标准溶液,按2.2.1项下工作条件,以稀释液为标准空白溶液,由石墨炉原子吸收分光光度计自动稀释并进行标准曲线测定,以各元素吸光度(Abs)为纵坐标、质量浓度(C)为横坐标绘制标准曲线;按2.2.1项下工作条件,以稀释液为空白,用原子荧光光度计进行测定,以相应元素的荧光强度为纵坐标(If)、质量浓度(C)为横坐标绘制标准曲线。2)LCP-MS法。取2.3.2项下系列混合标准溶液,按2.2.2 项下工作条件,以各元素质量浓度(C)为纵坐标、信号强度(I)为横坐标绘制标准曲线。结果见表3。
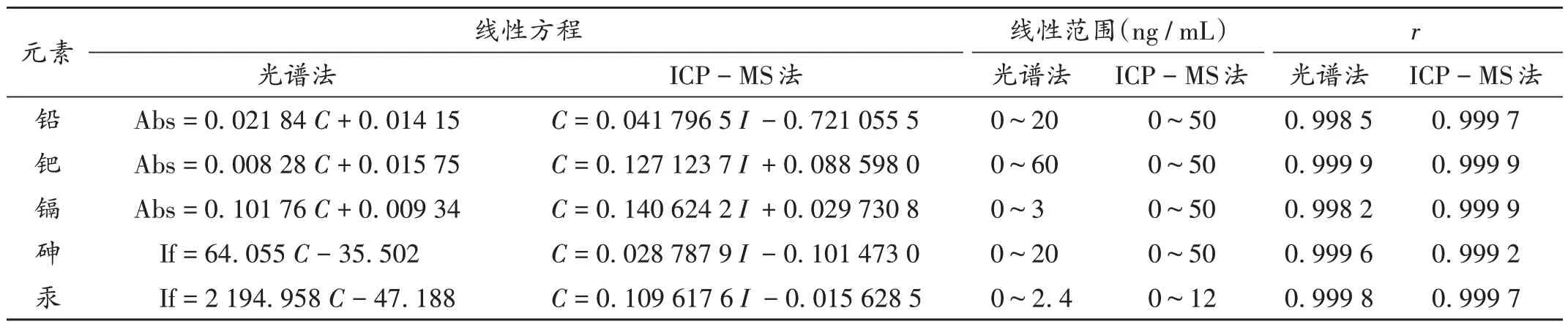
表3 线性关系考察结果Tab.3 Results of the linear relation test
检测限和定量限:1)光谱法。取2.3.2 项下供试品溶液,连续进样测定7 次,计算吸光度值的标准偏差(δ),按3.3δ与标准曲线方程斜率(S)的比值计算检测限;按10δ与标准曲线方程斜率的比值计算定量限[16]。2)ICP-MS 法。按取样量0.5 g,按稀释倍数25 倍计算,分别得方法检测限和方法定量限。结果见表4。
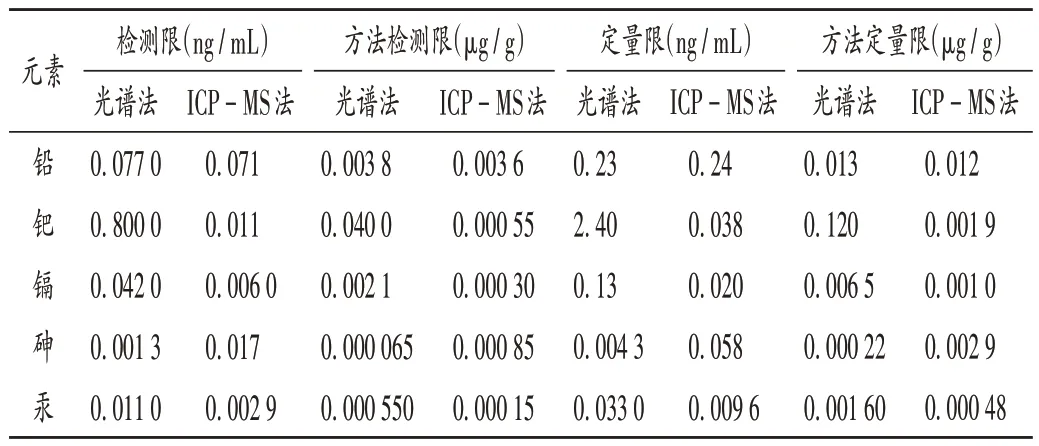
表4 检测限和定量限确定结果Tab.4 Determination results of LOD and LOQ
加样回收试验:分别吸取铅、钯单元素标准溶液各适量,用稀释液制成每1 mL 含铅1 000 ng、钯2 000 ng的铅、钯元素混合标准溶液。取盐酸羟考酮原料药(批号为1810001)9 份,每份0.5 g,精密称定,分别置微波消解罐中,加入硝酸5 mL、过氧化氢溶液2 mL,分别精密加入上述铅、钯元素混合标准溶液0.125,0.25,0.375 mL,各3 份,混匀,按2.3 项下方法制备加样供试品溶液。取加样供试品溶液和空白溶液,在相应元素条件下用石墨炉原子吸收分光光度计进行测定,根据标准曲线计算加样回收率,结果见表5。
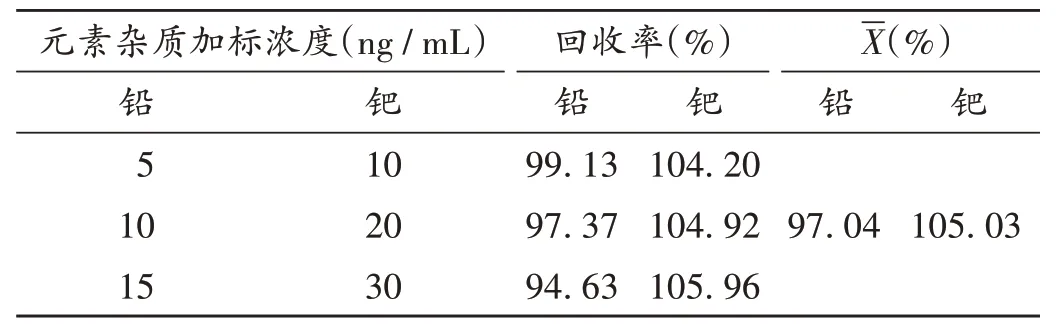
表5 铅和钯元素GF-AAS法加样回收试验结果(n=3)Tab.5 Results of the recovery test of lead and palladium by the GF-AAS method(n=3)
重复性试验:取盐酸羟考酮原料药(批号为1810001)6 份,每份0.5 g,精密称定,分别置微波消解罐中,加入硝酸5 mL、过氧化氢溶液2 mL,分别精密加入加样回收试验项下铅、钯元素混合标准溶液0.25 mL,按2.3 项下方法制备供试品溶液。结果铅和钯元素的加标浓度分别为10 ng/mL 和20 ng/mL,RSD分别为3.80%和2.10%(n=6)。
2.5 样品元素杂质测定
按2.3项下方法制备供试品溶液,对6个批次的盐酸羟考酮原料药和起始原料蒂巴因中铅、砷、汞、镉、钯元素分别进行光谱法和ICP-MS法检测,结果见表6和表7。
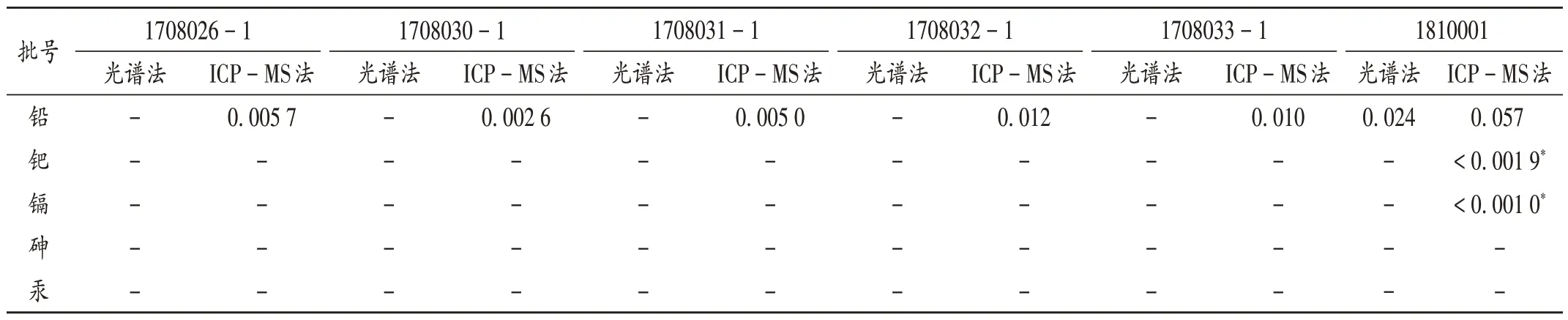
表6 盐酸羟考酮原料药中元素杂质测定结果(μg/g)Tab.6 Determination results of element impurities in the API of hydroxycodone hydrochloride(μg/g)
3 讨论
3.1 元素杂质控制种类选择
药品中的元素杂质对药品的安全性和有效性有显著影响,特别是重金属元素通常在人体难以代谢,易在体内蓄积,影响健康。各国药品监管机构对元素杂质的监管日趋严格和规范[17-20],元素杂质的控制和分析是药品研发的重点和难点,但元素杂质种类繁多,很难全部纳入质量标准。2020 年版《中国药典(二部)》中,盐酸羟考酮采用了重金属检查法对重金属限度进行控制,但方法专属性不强、灵敏度低,无法对药品中所有有害金属杂质进行有效控制。
本研究中通过对盐酸羟考酮原料药及其合成起始原料蒂巴因中ICH Q3D 规定的4种第一类元素,以及作为催化剂使用的钯元素进行了综合分析,其中铅元素在两者中均有检出,且合成起始原料蒂巴因中的检测结果高于盐酸羟考酮原料药,表明存在由原料引入铅元素的风险,故需对铅元素进行检测和控制。汞元素在合成起始原料和原料药中均未检出;砷元素在原料药中未检出;隔元素仅有1 批原料药中检出,但结果低于其定量限,故无需考虑将汞、砷、镉元素杂质列入原料药质量标准进行控制。另外,钯元素杂质是作为原料合成工艺的催化剂,且有1批原料药中检出钯,表明存在由生产工艺引入钯元素的风险。为保证原料药质量的安全性,需将铅、钯元素列入原料药质量标准进行控制。
3.2 元素杂质控制限度制订
ICH Q3D 中对铅元素杂质给出的最大日允许摄入量(PDE)为5 μg/d,钯元素为100 μg/d。对原料药的元素杂质的允许浓度按ICH Q3D 中PDE 值和浓度限度互相转换方法1进行简化计算,按日剂量不超过10 g/d的最大量,铅元素和钯元素控制限度分别为0.5 μg/g 和10.0 μg/g。
3.3 元素杂质控制方法选择
原子吸收法和ICP-MS 法均是目前非常成熟的金属杂质的检测方法。部分方法在建立时未经过综合研判,单纯追求使用高精尖设备,但更高端的设备往往普及率低,价格昂贵,使用、维护成本高。对于盐酸羟考酮原料药中金属杂质的控制,本研究中建立了微波消解法对样品进行前处理,分别使用GF-AAS法和ICP-MS法测定铅、钯元素,方法学验证结果表明均能满足2020年版《中国药典》9101 分析方法验证指导原则中的要求。GF-AAS法与ICP-MS法在测定铅元素时具有同量级的线性范围和检测限等。GF - AAS 法测定钯元素的检测限高于ICP-MS法,但检测结果远低于制订的控制限度。综合考虑,GF-AAS法的仪器普及率、操作和使用、维护成本均优于ICP-MS 法。故确定以GF-AAS 法作为盐酸羟考酮原料药元素杂质的控制方法。
