金属Al诱导Si晶化的Al/Si异质薄膜的原子输运模拟
2023-11-10谯进玉侯君祎严汝阳吴芳芳马晓波陈焕铭
侯 毅 王 宁 谯进玉 侯君祎 严汝阳 吴芳芳 马晓波 陈焕铭
(宁夏大学物理学院,宁夏 银川 750021)
0 引言
当金属与非晶半导体接触时,可以在较低温度下诱导非晶态的半导体晶化,这种现象称为金属诱导晶化(Metal-Induced Crystallization,MIC)[1]。非晶态材料为了启动晶化,必须要克服晶化能垒[2],所以需要很高的晶化温度。但是在金属和半导体接触时,金属的自由电子气延伸到与之接触的半导体材料中,对半导体的共价键产生库仑屏蔽效果,导致共价键弱化,使得半导体原子在一定范围内的迁移率提高,并在金属半导体接触面或是金属晶界处结晶。
对金属诱导晶化现象的研究,最早可以追溯到1969年。Oki等[3]发现,非晶Ge与Au、Ag、Cu、Al等金属接触时,最低晶化温度明显降低。Bosnell等[4]发现了金属与非晶Si接触时可以降低非晶Si的晶化温度。Herd 等[5]和Ottaviani 等[6]分别利用电子显微镜对上述现象进行了更细致的研究,并将其命名为金属接触诱导晶化。最初对金属诱导晶化现象的解释是在1973 年到1974 年间提出的。通过对试验现象的观察,Sigurd等[7]认为半导体材料的原子会先溶解到金属中,然后从金属中析出晶化的半导体。Brodsky和Turnbull则认为,相互接触的金属和半导体,在退火过程中会形成低温共熔体。在共熔体中,半导体材料的晶化温度降低,导致产生金属能够在较低温度状态下诱导半导体材料结晶的现象。直到将原位加热透射电子显微镜技术应用到金属诱导晶化的研究中,人们才发现金属诱导晶化过程中并没有液相的金属或半导体存在,而是完全的固-固相变过程。
按照是否有金属-半导体化合物的生成,金属诱导晶化被分为两大类。没有金属-半导体化合物生成的,包括Al、Au、Ag 等金属与非晶Si、Ge 构成的体系[8-10];有金属-半导体化合物生成的,包括Ni、Pd、Cu等金属与非晶Si、Ge构成的体系[11-13]。没有金属-半导体化合物生成的体系,晶化温度较低,并且通常伴随着半导体和金属膜层的翻转。有金属-半导体化合物生成的体系,晶化温度较高,且多有金属-半导体化合物的形成。一般情况下,Si的晶化温度高于700 ℃,但是在诱导金属的作用下,这一温度大幅降至200 ℃左右,极大地扩展了晶体Si薄膜的应用场景。
本研究针对以上述及的研究与学术背景,对Al/Si 异质非晶薄膜在热处理过程中Si 的晶化过程进行理论计算与模拟。从原子层次分析了金属Al诱导Si晶化的物理过程与机理,为后续研究,特别是热处理工艺的制定等方面提供预测和借鉴。
1 计算模型
Al/Si薄膜分子动力学计算的初始结构如图1 所示。模型的最下层为α石英,其晶格常数a=b=5.03 Å,α=β=90°,Γ=120°,中间层为非晶态的Al层,最上层为非晶态的Si层。为了模拟Al层和Si层界面附近Si一侧的电荷密度分布,同时考虑到Al 原子在Si 材料中库仑屏蔽效应作用的距离有限,选择Si-I晶胞为本征模型,计算了晶胞上表面的Si 原子替换为Al 原子时电荷密度分布的变化。两个模型的X、Y方向应用周期性边界条件,Z轴方向上添加了厚度为20 Å的真空层,以避免周期性相互作用的影响。
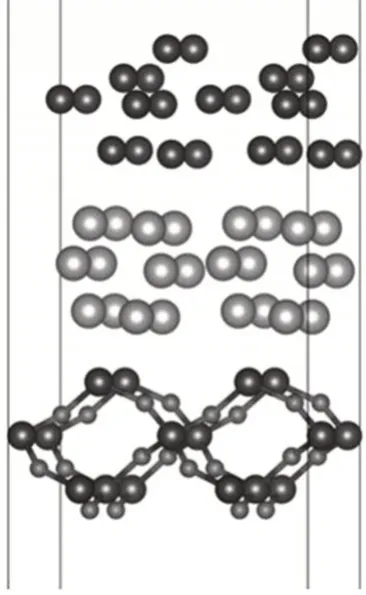
图1 以石英为衬底的Al/Si异质薄膜的初始结构
2 计算方法
计算采用第一性原理计算软件VASP 软件包[14],体系离子实与价电子之间的相互作用采用PAW方法进行描述。平面波的截断能设置为300 eV,交换关联泛函采用PBE 的GGA 广义梯度算法[15]。弛豫过程中能量收敛标准为1×10-5eV。基于NVT系综对图1 所示结构进行分子动力学模拟。计算过程中固定α 石英层的结构,对Al、Si 层的原子进行弛豫。起始和结束温度均设置为1 073 K,步长为3 fs,弛豫过程共6 000 步。计算过程中布里渊区k 点网格采样选用Monkhorst-Pack 方法[16],晶体的轨道哈密顿布居(COHP)及其积分(ICOHP)利用Lobster 软件[17-18]进行计算。
3 结果与讨论
为了从理论上分析试验中观察到的Al/Si 异质膜层翻转的现象,采用分子动力学方法对所建立的Al/Si 异质薄膜进行热处理,并分析热处理过程中Al、Si原子的输运情况。在Al/Si薄膜热处理过程中,系统温度及系统总能随时间的变化分别如图2(a)和图2(b)所示,由图可知,热处理过程中,系统温度始终在1 073 K 上下浮动。系统总能在最初的500 步内下降明显,随着热处理时间的延长,总能呈现波动下降的趋势。以α石英为衬底的Al/Si薄膜在热处理之前及热处理6 000 fs、12 000 fs、18 000 fs 后的结构如图3 所示。从结构演变的结果可知,热处理过程中,Al 层和Si 层中的原子彼此间相互扩散。随着热处理时间的延长,Al原子逐渐向上移动,Si原子逐渐向下移动,使得膜层系统进入能量更低的较为稳定的状态,并开始出现膜层翻转的趋势。
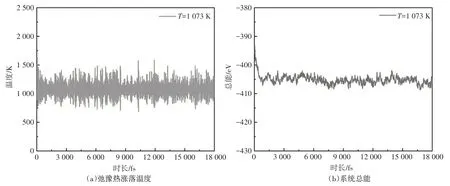
图2 Al/Si异质薄膜的原子弛豫热涨落温度及总能随时间的变化

图3 分子动力学模拟过程中的原子弛豫过程
Al/Si界面处的电荷密度分布如图4所示。与图4(a)相比,图4(a)中在Al原子取代了最上层的Si原子之后,剩余Si原子之间的电荷密度明显降低,且距离Al 原子越近,电荷密度衰减得越明显。为了进一步确认引入Al 原子之后Si 原子间成键情况的变化,计算了如图4(a)和图4(b)两种情况下处于同一相对位置的Si 原子对Si3-Si8 和Si3-Si6 的晶体轨道哈密顿布居(COHP)及其积分(ICOHP),如图5 所示。从-COHP的曲线可知,引入Al原子前后,两个Si原子之间的成键状态有所变化。通过-COHP的积分值能够更直观地看出,在引入Al原子之后,两个Si原子之间的轨道相互作用明显减弱。说明在Al诱导非晶Si晶化的过程中,在靠近Al原子一定距离范围内,由于Al原子的库仑屏蔽作用使得Si—Si键的强度减弱,有利于Si原子的迁移,使得非晶Si的晶化能垒降低。
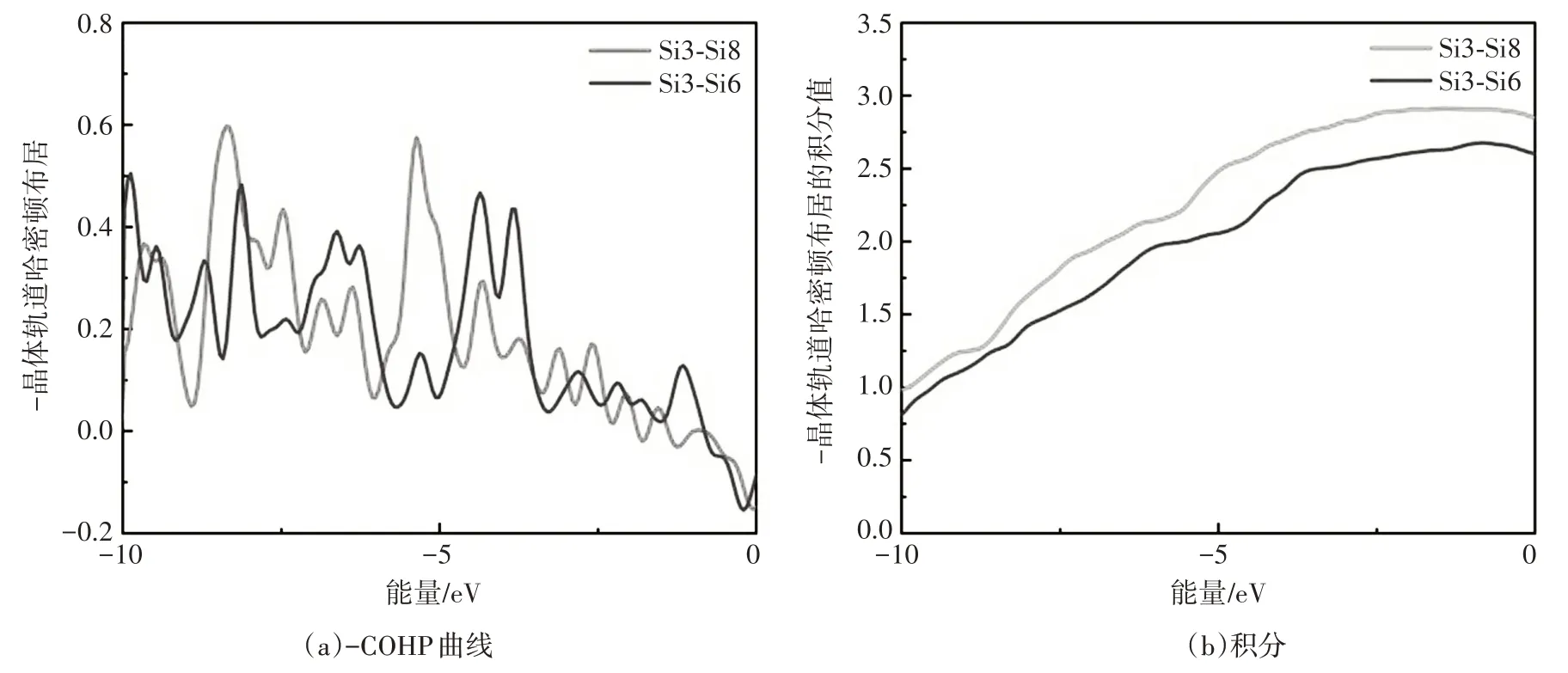
图5 Si原子间的-COHP曲线及其积分
4 结论
①Al/Si异质薄膜在热处理过程中,Al、Si原子发生了互扩散现象。随热处理时间的延长,Al 原子逐步上移,Si原子逐步下移,膜层系统进入能量更低的较为稳定的状态。Al/Si 膜层在结构演变的过程中逐渐出现膜层翻转的趋势。
②Al原子的引入导致Si原子间成键状态发生变化。Al原子的库仑屏蔽作用使得Si—Si键的强度减弱,有利于Si原子的迁移及非晶Si晶化温度的降低。
