李白盾蚧转录组分析及SSR位点开发
2023-11-09杜炫星牛敏敏卢运运魏久锋张虎芳
杜炫星,牛敏敏,赵 清,蔡 波,卢运运,魏久锋*,张虎芳,2*
(1.山西农业大学植物保护学院,山西太谷030801;2.忻州师范学院生物系,山西忻州 034000;3. 海口海关热带植物隔离检疫中心,海口570311)
李白盾蚧Pseudaulacaspisprunicola(Maskell),隶属于半翅目Hemiptera盾蚧科Diaspididae白盾蚧属Pseudaulacaspis,与桑白盾蚧Pseudaulacaspispentagona在形态上非常相似。汤祊德先生在《中国园林主要蚧虫·第三卷》中认为李白盾蚧P.prunicola为桑白盾蚧P.pentagona的同种异名,但Miller和Davidson等人研究认为P.prunicola和P.pentagona为两个不同的物种,形态上主要区别点为雌成虫臀末三个侧叶间的腺刺,P.prunicola成双而端尖,P.pentagona单一而端部分叉,2019年Normark等人通过多基因联合分析表明P.prunicola和P.pentagona不是同一物种。李白盾蚧广泛分布于日本、韩国、印度和美国,2014年首次被深圳出入境检验检疫局截获,在我国的宁夏和台湾局部分布,是一种重要的入侵生物(向才玉等,2015)。该虫寄主植物范围广泛,主要危害李、桃、樱桃等植物,同时对多种经济林木和观赏植物都能造成严重危害。由于李白盾蚧与桑白盾蚧在我国经常被认为是一个物种,所以迄今为止其在我国没有准确的分布与危害情况,但经笔者采集调查发现其在我国很多省份都已经大量发生,严重危害我国果树生产及园林绿化。
转录组测序是指利用第二代高通量测序技术进行cDNA测序,能全面快速地获取研究材料特定组织在某一状态下全部转录本的序列信息和表达信息(胡俊杰等,2017)。Vera等(2008)利用罗氏454测序技术进行庆网蛱蝶Melitaeacinxia的转录组测序,是首个采用从头组装的无参昆虫转录组研究,由此拉开了昆虫转录组研究的序幕。在蚧虫中进行转录组分析的物种也有很多,例如:中华紫胶虫Kerriachinensis(Wangetal.,2019)、曼粉蚧Maconellicoccushirsutus(Kohlietal.,2021)、白蜡虫Ericeruspela(Yangetal.,2015)、扶桑绵粉蚧Phenacoccussolenopsis(胡俊杰等,2016)及菠萝洁粉蚧Dysmicoccusbrevipes(何衍彪等,2019)等。
简单重复序列(simple sequence repeat, SSR),又被称为微卫星(microsatellite)DNA,广泛分布于真核生物基因组中,由若干个碱基大小为1~6 bp的简单重复单元串联组成(孙荆涛等,2012;刘倩倩等,2021)。SSR标记具有种类多、重复性高、共显性遗传、多态性信息丰富、操作简易等特点,被广泛应用于分子遗传育种、种群遗传多样性分析、近缘物种鉴定、基因定位与克隆、遗传连锁图谱构建等各个领域(Zonneveldetal.,2012;Tangetal.,2015;Zhuetal.,2019)。但传统的SSR分子标记开发方法实验步骤复杂、效率低、重复性差(Roderetal.,1998;Uzunova and Ecke,1999)。高通量测序技术和生物信息学的快速发展,为SSR标记的开发提供了更加高效、便捷的方法(Schoebeletal.,2013)。目前,基于转录组数据库已成功挖掘出了印度谷螟Plodiainterpunctella(唐培安等,2016)、沙棘木蠹蛾Eogystiahippophaecolus(崔明明等,2017)、东方粘虫Mythimnaseparata(Walker)(李微等,2017)、梨小食心虫Grapholithamolesta(冷春蒙,2018)、星天牛Anoplophorachinensis(韩小红等,2019)、椰心叶甲啮小蜂Tetrastichusbrontispae(刘华伟等,2021)、灰茶尺蠖Ectropisgrisescens(王定锋等,2021)、温带臭虫Cimexlectularius(李敏等,2019)等多种昆虫的SSR位点,为其今后种群遗传结构、发生动态及扩散历史研究提供依据。
迄今为止,对李白盾蚧的研究大多集中在其生物学习性以及化学防治等方面(Uchidaetal., 2021),而对其分子标记的研究还处于空白状态。本研究利用转录组测序技术,结合生物信息学方法,对李白盾蚧的SSR位点进行挖掘,并进行序列特征的统计与分析,设计和筛选能够稳定扩增的SSR引物,为李白盾蚧的分子鉴定与遗传结构分析奠定基础。
1 材料与方法
1.1 供试昆虫
本研究试虫采集自山西省晋中市太谷区西山底村(37°20′59.4″N,112°32′54.5"E)。设3个重复组,每组30头雌成虫,经液氮速冻后,在-80℃超低温冰箱中保存。用于验证SSR引物的李白盾蚧雌成虫采集信息见表1,所有样本均浸泡于无水乙醇中,在-20℃低温冰箱保存。使用Ezup柱式动物基因组DNA抽提试剂盒(生工生物工程(上海)有限公司)进行总DNA提取,-20℃保存。上述所有样本均提取线粒体COI同NCBI数据库比对进行分子鉴定,结果为李白盾蚧。

表1 李白盾蚧6个地理种群样本信息
1.2 总RNA提取、cDNA文库构建及测序
委托北京诺禾致源科技股份有限公司进行转录组测序。首先用Trizol法提取李白盾蚧的总RNA,RNA质量以紫外分光光度计和琼脂糖凝胶电泳检测。样品检测合格后,通过Oligo(dT)磁珠富集带有polyA尾的mRNA,随后加入Fragmentation Buffer将得到的mRNA随机打断。以片段化的mRNA模板,随机寡核苷酸为引物,在M-MuLV逆转录酶体系中合成cDNA第一条链,随后RNaseH降解RNA链,并在DNApolymerase I体系下,以dNTPs为原料合成cDNA第二条链。纯化后的双链cDNA经过末端修复,加A尾并连接测序接头,用AMPure XP beads筛选370~420 bp的cDNA,进行PCR扩增并再次使用AMPure XPbeads纯化PCR产物,最终获得文库。文库构建完成后,使用Agilent 2100 bioanalyzer对文库质量进行评估,合格后利用Illumina NovaSeq 6000(Illumina,USA)平台进行测序。
1.3 原始数据的拼接和de novo组装
对李白盾蚧进行转录组测序得到原始序列(Raw reads),通过去除带接头的读数、去除含N(N表示无法确定碱基信息)的读数、去除低质量读数,得到高质量序列(Clean reads)。同时对clean reads进行Q20(碱基正确识别率达到99%以上的碱基占比)、Q30(碱基正确识别率达到99.9%以上的碱基占比)和GC含量的计算。利用Trinity v2.6.6(Grabherretal.,2011)软件对clean reads进行组装拼接,获得转录本序列。原始数据已上传至NCBI数据库中,登录号为PRJNA863380。
1.4 李白盾蚧转录组unigenes的功能注释
在Trinity拼接的基础上,使用Corset v4.6(Davidson and Oshlack,2014)软件进行转录本聚类,去除冗余转录本,得到非冗余unigenes。将所有unigenes使用(Diamond v0.8.22、blast2go v2.5等软件,以及KAAS在线网站等)软件同Nr、Nt、Swiss-prot、Pfam、KOG、KEGG和GO数据库比对,进行基因功能注释。
1.5 SSR位点筛选与引物鉴定
使用软件MISA v1.0(Thieletal.,2003)对李白盾蚧unigenes进行SSR位点搜索,搜索标准为:重复单元长度大小依次为1、2、3、4、5和6 bp,最小重复次数分别为10、6、5、5、5和5,SSR位点侧翼序列长度大于100 bp。
根据MISA的输出结果,使用Primer Primer 3 v2.3.5(Untergasseretal.,2012)软件批量设计引物,遵循以下原则对所有引物进行初步筛选:(1)GC含量为40%~60%;(2)退火温度为50~60℃,正、反向引物退火温度相差不超过5℃;(3)引物长度建议为17~28 bp,最好不要超过30 bp;(4)预期PCR产物大小为150~300 bp;(5)避免引物二聚体和发夹结构。经初步筛选后,随机挑选50对引物,由生工生物工程(上海)有限公司进行合成。以李白盾蚧总DNA为模板进行PCR反应,反应体系20 μL:DNA模板1 μL、2×Taq PCR Master Mix 10 μL、上下游引物(10 μmol/L)各1 μL、ddH2O补足至20 μL。反应程序为:94℃预变性3 min、94℃变性30 s、Tm复性30 s,72℃延伸30 s,共35个循环;72℃延伸5 min。PCR产物经1%琼脂糖凝胶电泳检测。
筛选引物时,首先使用宁夏银川的1个样本总DNA对所有的50对引物进行PCR反应和琼脂糖凝胶电泳检测,其中34对引物PCR扩增电泳检测的条带单一、清晰可见,与目的片段大小基本一致,其余16对引物没有片段,或者与目的片段大小不一致。将上述34对引物继续用6个地理种群(表1)每个种群3个样本的总DNA进行PCR扩增稳定性检测。上述所有样本均为雌蚧虫。
2 结果与分析
2.1 李白盾蚧转录组测序、拼接和组装
运用Illumina NovaSeq 6000测序平台对李白盾蚧转录组测序,平均获得22 348 052条原始序列。经数据过滤后,平均得到21 570 143条clean reads,碱基错误率为0.03%,每组样本均生成6 Gb以上的高质量读数,Q20平均为97.01%,Q30平均为91.95%,GC含量平均为43.63%,测序质量良好。共组装到60 296条转录本,24 967条unigenes,总长度为41 668 408bp,最大长度为28 540 bp,平均长度为1 669 bp。N50和N90分别为2 816 bp和646 bp,拼接效果良好。组装得到的24 967条unigenes全部用于后续SSR位点搜索。
2.2 李白盾蚧转录组unigenes的功能注释
通过Nr、Nt、Swiss-prot、Pfam、KOG、KEGG和GO七大数据库对李白盾蚧24 967条unigenes进行注释,其中注释到Nr数据库的unigenes最多,为11 244(45.03%)条;2 172(8.69%)条unigenes注释到NT数据库;6 225(24.93%)条unigenes注释到KO数据库;9 417(37.71%)条unigenes注释到Swiss-prot数据库;11 122(44.54%)条unigenes注释到Pfam数据库;11 121(44.54%)条unigenes注释到GO数据库;5 805(23.25%)条unigenes注释到KOG数据库。所有数据库都能注释到的unigenes有1 290条,占5.16%;在至少一个数据库中注释到的unigenes有13 541条,占54.23%(表2)。
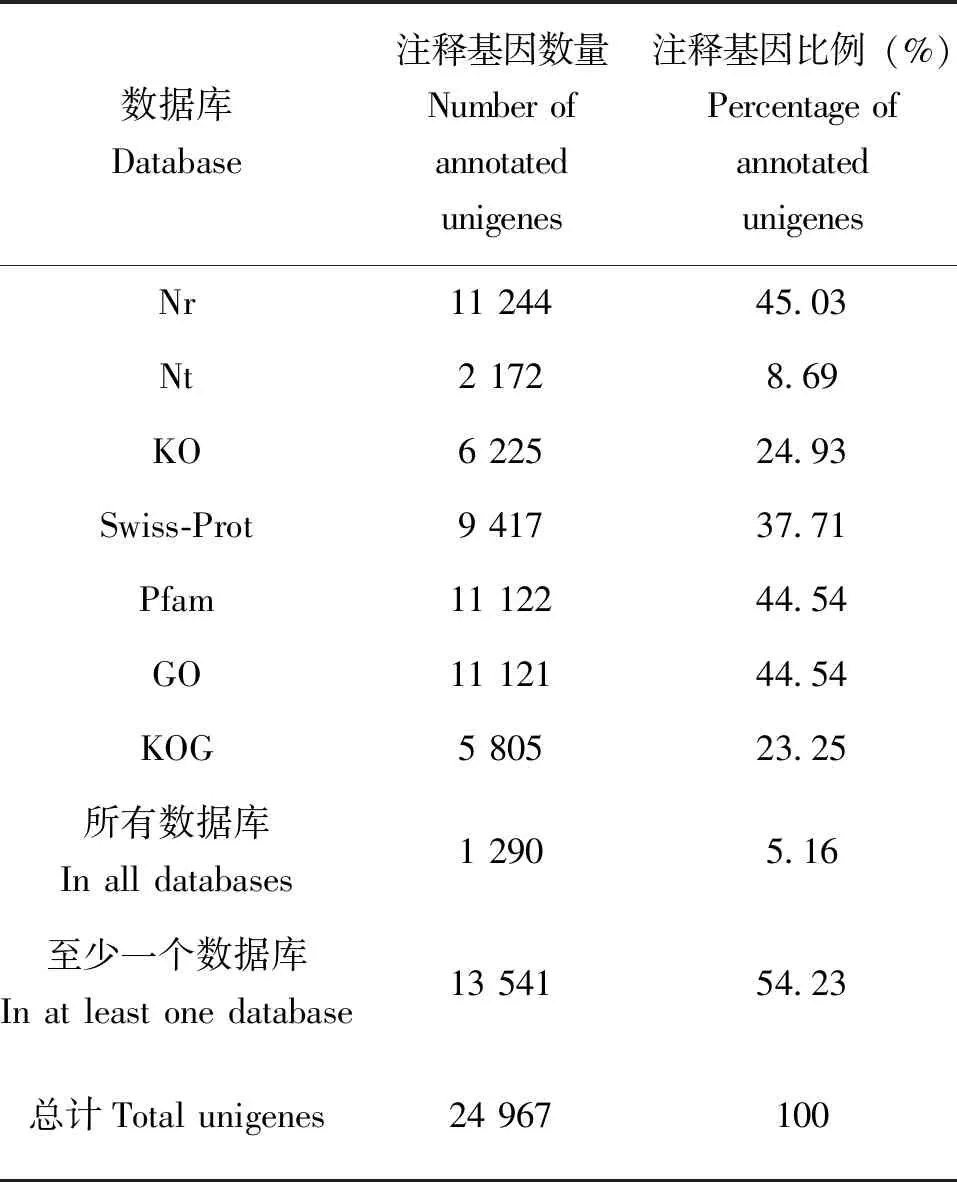
表2 李白盾蚧转录组unigenes在7个数据库中的注释结果统计
通过与Nr数据库进行同源性比对注释,李白盾蚧转录组注释到其他物种的unigenes共11 244条。其中与烟粉虱Bemisiatabaci的相似性序列最多,所占比例为13.3%;其次为塞古都斯白蚁Crytotermessecundus,所占比例为6.7%;内华达白蚁Zootermopsisnevadensis、灰飞虱Laodelphaxstriatellus和褐飞虱Nilaparvatalugens所占比例分别为4.7%、4.5%和4.8%,其他物种所占比例为66.0%(图1)。
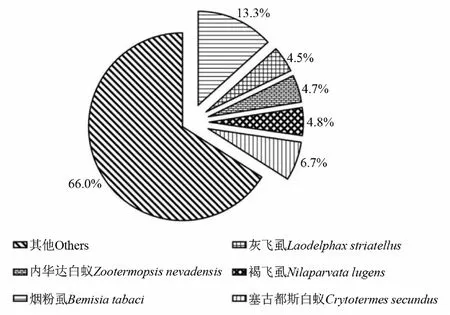
图1 李白盾蚧与其他物种Nr数据库转录组数据比对相似度
利用Blast2go(Götzetal.,2008)软件对李白盾蚧转录组11 121条unigenes进行GO注释,得到49 745条功能注释(图2),分为三大类:生物学进程(Biological process)、细胞组分(Cellular component)和分子功能(Molecular function)。生物学进程中共注释到24 822条序列,分为24个次级功能条目,其中细胞进程(Cellular process)数量最多,为6 611条;其次为代谢进程(Metabolic process)、生物调节(Biological regulation)、生物进程调控(Regulation of biological process)和定位(Localization),数量分别为5 647条、2 580条、2 408条和2 138条。在细胞组分中共有10 962条功能注释,其中细胞结构体(Cellular anatomical entity)数量最多,为5 178条,其次为细胞内(Intracellular),2 924条。分子功能分类中共注释到13 961条序列,其中结合(Binding)6 365条、催化活性(Catalytic activity)4 675条。
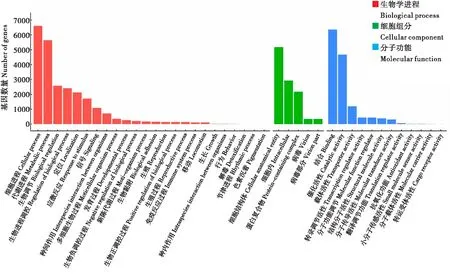
图2 李白盾蚧unigenes GO功能注释
通过对李白盾蚧转录组24 967条unigenes进行KOG直系同源分类,共得到5 805条基因注释,分为25类,分类结果见图3。其中,R类一般功能预测(General function prediction only)和T类信号转导相关(Signal transduction mechanisms)的基因条数最多,分别为992条和748条;其次为O类翻译后修饰(Posttranslational modification)、蛋白质周转(Protein turnover)和分子伴侣(Chaperones)相关共633条,K类转录相关(Transcription)417条。除有具体功能注释外还有S类未知功能(Function unknown)419条。
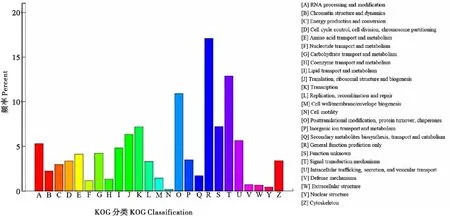
图3 李白盾蚧unigenes KOG分类注释
通过对24 967条unigenes进行KEGG代谢通路富集分析,结果如图4所示,共有6 668条unigenes参与到细胞进程(Cellular processes)、环境信息处理(Environmental information processing)、遗传信息处理(Genetic information processing)、新陈代谢(Metabolism)和有机系统(Organismal systems)这五大类生物代谢途径中。其中有机系统途径中基因最多,为1 662条,主要参与内分泌系统(Endocrine system)、免疫系统(Immune system)和神经系统(Ervous system)等过程,数量分别为390条、283条和224条。在所有的34组次级代谢途径中,信号转导(Signal transduction)途径的基因最多,为836条。这些与代谢途径相关的基因分布于已知的280个代谢通路中,其中富集最多的10条通路分别是内质网中的蛋白质加工(Protein processing in endoplasmic reticulum)、核糖体(Ribosome)、RNA运输(RNA transport)、剪接体(Spliceosome)、内吞作用(Endocytosis)、碳代谢(Endocytosis)嘌呤代谢(Purine metabolism)、溶酶体(Lysosome)、cAMP信号传导途径(cAMP signaling pathway)和细胞周期(Cell cycle)。
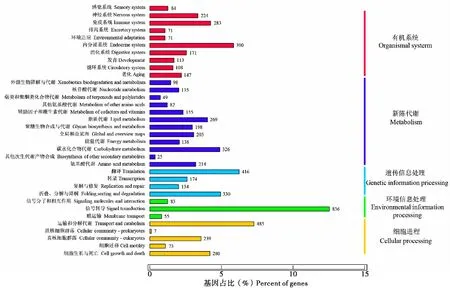
图4 李白盾蚧unigenes KEGG通路分析
2.3 SSR位点的数量和分布特征
从李白盾蚧24 967条unigenes数据中,共检测出18 193个SSR位点,分布在9 043条unigenes中,其中3 861条unigenes含有一个以上的SSR位点,复合SSR位点1 704个。SSR位点在转录组数据中的出现频率(含SSR位点的unigene数量/总unigene数量)为36.22%,SSR位点的分布频率为(SSR位点个数/总unigene数量)72.87%,平均每2.29 Kb发现一个SSR位点。
SSR位点中,重复基序为单核苷酸的位点所占比例最高,为72.03%,其次为二核苷酸、三核苷酸和四核苷酸,分别为8.48%、15.90%、3.10%(表3)。单核苷酸重复中,A/T为主要的重复基序,占SSR位点总数的71.16%;二核苷酸重复中,AG/CT和AC/GT为主要的重复类型,分别占总数的5.20%和1.83%;三核苷酸重复中,数量最多的重复基序为AAT/ATT和ACG/CGT,所占比例分别为5.10%和3.22%;四核苷酸、五核苷酸和六核苷酸,共占SSR位点总数的7.55%(图5)。

图5 基于李白盾蚧转录组数据的不同SSR位点重复基序频率分布图
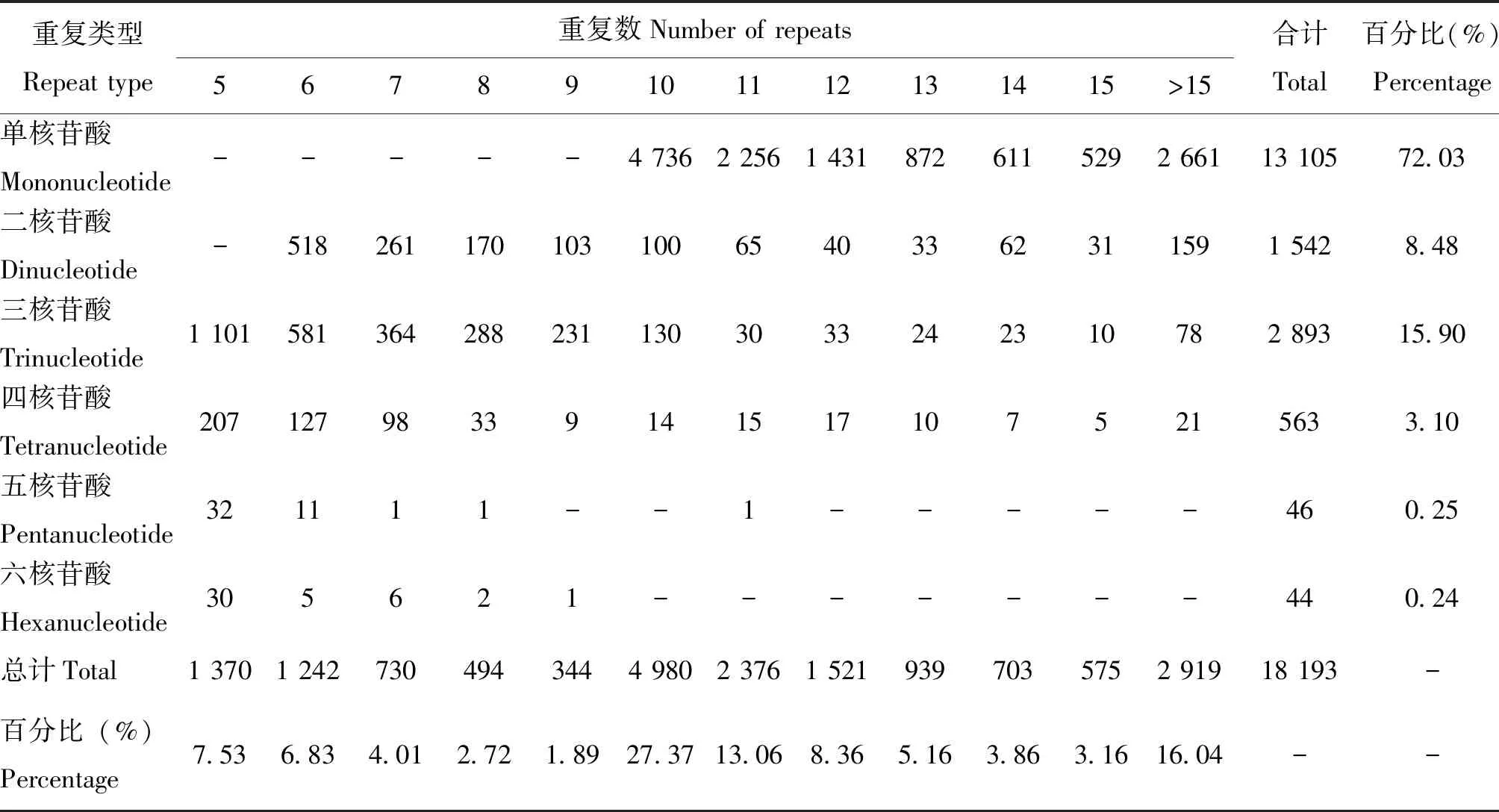
表3 基于李白盾蚧转录组数据的不同类型SSR位点数量统计
李白盾蚧转录组数据中,SSR位点的重复次数集中在5~15次,所占比例为83.96%;其余大于15次大多为单核苷酸和二核苷酸重复,少部分为三核苷酸和四核苷酸重复,共占总数16.04%。单核苷酸重复次数最多为10次,二核苷酸为6次,其余重复类型重复次数最多为5次(表3)。
2.4 SSR位点的长度分布
由于部分SSR位点为复合型,所以长度分析中微卫星的总数为16 087个。在所有的微卫星中,长度为10~469 bp不等,平均长度为22.98 bp,序列长度为10~15 bp的共有9 603个,占总SSR个数的59.69%,其中单核苷酸、二核苷酸、三核苷酸重复单元分别占85.29%、5.96%、8.76%;序列长度为16~20 bp的共有1 956个,占总SSR个数的12.16%,其中单核苷酸占比最多(55.62%),其次为三核苷酸(22.34%);序列长度为21~25 bp的共有1 338个,占SSR总数的8.32%,其中单核苷酸和三核苷酸占比相近,分别为38.57%和37.37%,复合型、二核苷酸和四核苷酸占比相近,五核苷酸最少为1.79%;序列长度大于25 bp的共有3 190个,占总SSR个数的19.83%,其中1~6核苷酸重复和复合型都有,最多的为复合型(52.51%)(图6)。
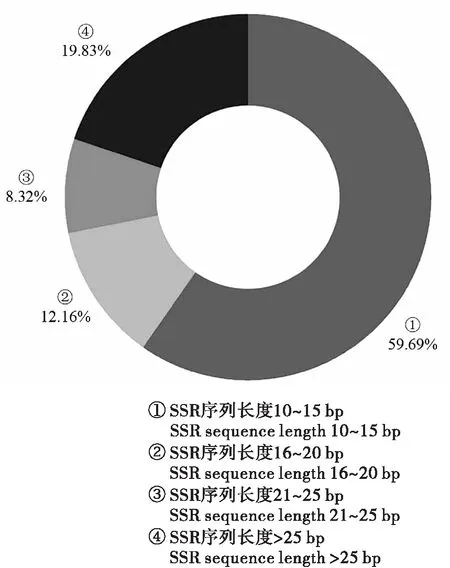
图6 李白盾蚧转录组SSR的长度分布
2.5 李白盾蚧SSR引物设计与验证
基于已筛选出的李白盾蚧SSR位点,利用Primer Primer 3软件进行引物批量设计,共有12 538个位点设计出了引物。根据上述引物筛选原则,对所有的12 538对引物进行筛选过后,共有305对符合条件,从中随机挑选50对引物进行合成。
首先利用宁夏银川的单头李白盾蚧DNA样本对50对引物进行PCR扩增,共有34对引物成功扩增出与预期片段大小一致的特异性片段,条带清晰明亮(表4,图7)。继续用6个不同地理种群李白盾蚧DNA样本对上述成功扩增的34对引物进行扩增稳定性验证,结果PP_4、PP_8、PP_31、PP_39在海南海口种群中无扩增条带,或条带模糊,引物PP_38在贵州雷山种群的扩增条带与目的条带大小不一致(图8),剩余29对引物可以稳定扩增出目的片段,且与预期目的片段大小一致,有效扩增率达到58%。引物的扩增多态性仍需进一步验证。已成功扩增的SSR引物可用于李白盾蚧进一步的遗传多样性研究。
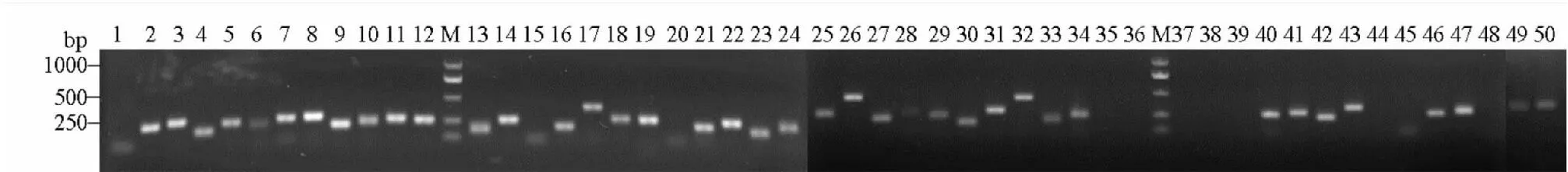
图7 李白盾蚧宁夏银川种群50个SSR位点扩增电泳图
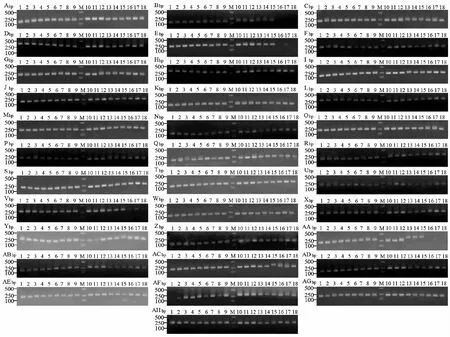
图8 李白盾蚧6个地理种群3个不同DNA样本34个SSR位点扩增电泳图
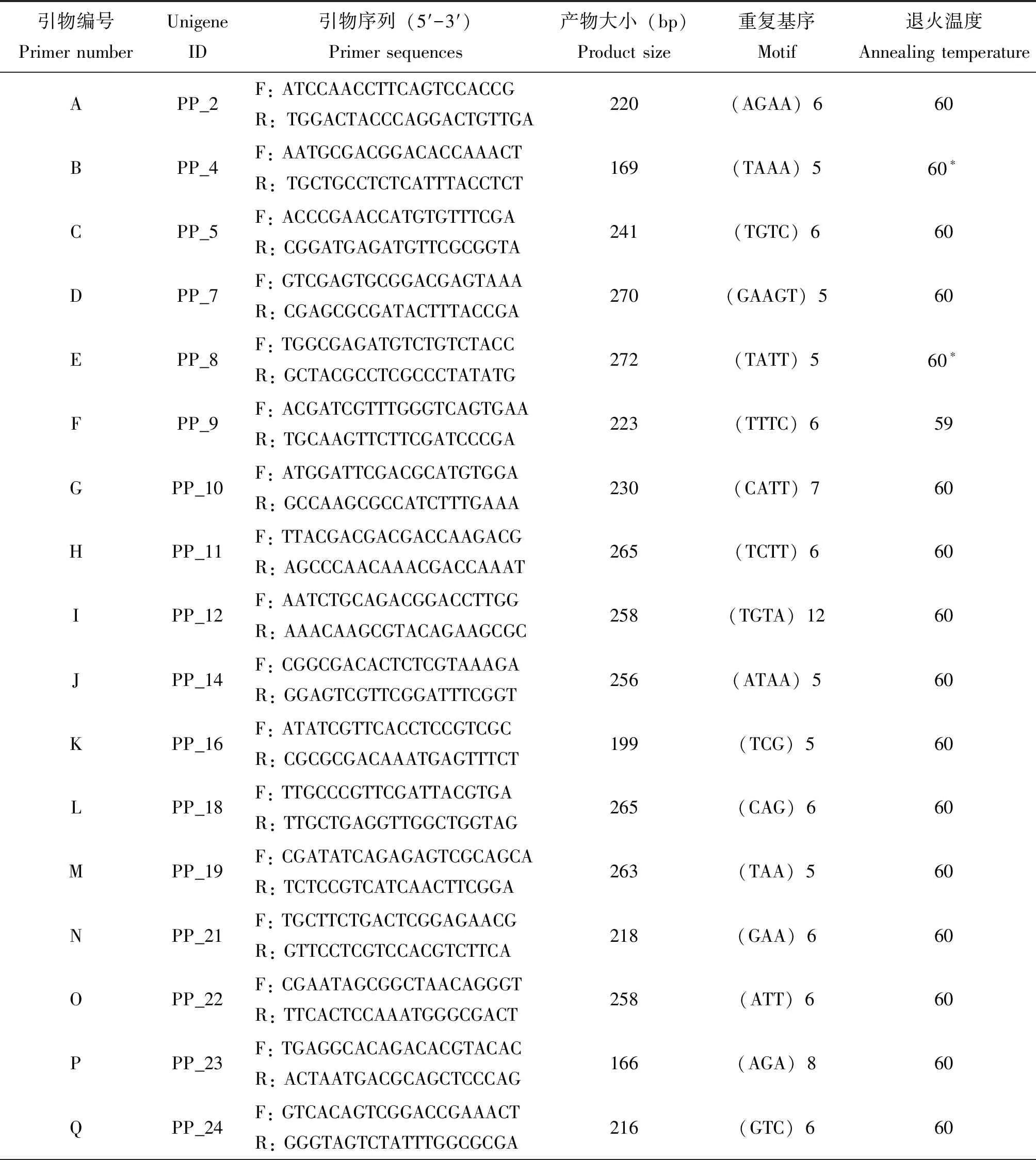
表4 李白盾蚧SSR引物信息
3 结论与讨论
随着新一代高通量测序技术的发展,越来越多昆虫的转录组数据以及全基因组序列被公布,但盾蚧科昆虫迄今为止还没有已公布的全基因组序列,转录组数据也少之又少,导致关于盾蚧科昆虫的基因功能以及遗传进化相关研究存在一定局限。本研究对李白盾蚧转录组数据进行测序、组装与分析,共得到60 296条转录本,24 967条unigenes,所有样本的Q30比例均不低于91%,N50为2 816 bp。一般认为Q30比例大于80%,则测序质量可靠(王兴春等,2015);N50长度大于800 bp,则说明组装得到的序列完整性较高(胡俊杰等,2017)。由此可见本试验所获得的转录组数据质量较好,序列拼接的完整性较好,保证了后续转录组分析的准确性。
通过与Nr和Swiss-prot蛋白质数据库进行比对,24 967条unigenes中共有11 244条(45.03%)注释到Nr数据库,9 417条(37.71%)注释到Swiss-prot数据库中,说明本次转录组测序获得了大量在李白盾蚧中表达的不同基因。GO分类注释是一套国际标准的基因功能分类系统,分为生物学过程、分子功能和细胞组分三大类,分别用来描述基因编码的产物所参与的生物过程、所具有的分子功能及所处的细胞环境。本研究GO分类注释结果中,共得到49 745条注释结果,其中生物学进程数量最多占主导地位,其次为分子功能,细胞组分注释结果最少(图3),这与已报道的例如中华大仰蝽Notonectachinensi(李敏等,2022)、春尺蠖Apocheimacinerarius(陈龙等,2022)、花椒窄吉丁Agriluszanthoxylumi(巩雪芳等,2020)、阿尔泰蝠蛾Hepialusaltaicola(孙涛等,2021)等生物学进程最多,细胞组分次之,分子功能最少的昆虫不同,这可能是由于不同昆虫在长期进化过程中所产生的差异。通过KOG直系同源分类注释,李白盾蚧转录组中与一般功能预测和信号转导相关的unigenes数目最多,KEGG代谢通路分类中共获得280条代谢通路,其中与有机系统通路相关的基因数目最多。上述结果可以为后续大量挖掘李白盾蚧生长发育过程中的重要表达基因、基因克隆等提供可靠的分子依据。
本研究基于李白盾蚧转录组数据库24 967条unigenes,成功挖掘出18 193个SSR位点,分布在9 043条unigenes中,平均每2.29 Kb发现一个SSR位点,发生频率为36.22%,高于许多已报道的昆虫包括意大利飞蝗Calliptamusitalicus(17.58%)(桑迪等,2020)、黄地老虎Agrotissegetum(6.09%)(常虹等,2022)、星天牛(18.85%)(韩小红等,2019)、扶桑绵粉蚧(5.79%)(罗梅等,2014)、灰茶尺蠖(20.26%)(王定锋等,2021)、窄足真蚋Simulium(Eusimulium)angustipes(36.05%)(郭欢等,2018),但低于梨网蝽Stephanitisnashi(40.37%)(谢瑾燕等,2019)、椰心叶甲啮小蜂(39.96%)(刘华伟等,2021)等。由于本研究将单核苷酸重复类型计入SSR位点总数,所以其发生频率较高,但例如齿缘刺猎蝽Sclominaerinacea(4.99%)(黎东海和赵萍,2018)、中华大仰蝽(7.07%)(李敏等,2022)等并没有将单核苷酸重复类型计入SSR位点总数中,所以其发生频率相对较低。不同昆虫间SSR位点的发生频率不同,究其原因可能与物种本身的差异有关,同时,转录组数据库大小、筛选SSR位点的标准、测序时RNA的质量等都有可能是不同物种SSR发生频率出现差异的原因。
本研究中,李白盾蚧转录组挖掘出的微卫星位点种类丰富,1~6核苷酸重复类型均有出现,其中数量最多的为单核苷酸重复类型,占总数的72.03%,其次为三核苷酸重复类型,占总数的15.90%,这一结果同许多已报道的昆虫相似,例如沙葱萤叶甲Galerucadaurica(张鹏飞等,2016)、窄足真蚋(郭欢等,2018)、黑腹胃蝇Gasterophiluspecorum(陈亘浓等,2018)等。而不同于沟眶象Eucryptorrhynchuschinensis(武政梅等,2016)、二点委夜蛾Athetislepigone(Lietal.,2013)、白背飞虱Sogatellafurcifera(Xuetal.,2012)、云南切梢小蠹Tomicusyunnanensis(袁远等,2014)等以三核苷酸重复类型为主的昆虫,其原因可能是SSR位点的筛选标准不同,本研究中单核苷酸重复类型重复次数最少为10次,但上述以三核苷酸重复类型为主的昆虫,其单核苷酸重复类型重复次数为12次或12次以上,所以其单核苷酸重复类型的数目相对较少,三核苷酸重复类型数目较多。符合遗传编码过程中,三联体密码子的重要性。
在李白盾蚧SSR所有核苷酸重复基序中,A/T所占比例为71.16%,数量最多,与已报道的大多数昆虫分析结果一致,例如印度谷螟(唐培安等,2017)、褐飞虱(刘玉娣和侯茂林,2010)、扶桑绵粉蚧(罗梅等,2014)等。二核苷酸重复中GC/GC的数量最少,仅有21个,占总数的0.21%,但在印度谷螟、二点委夜蛾、粘虫Mythimnaseparate(Walker)(胡艳华等,2015)等鳞翅目昆虫中则结果不同。据报道,动物SSR中的GC/GC含量较低甚至没有(Duanetal.,2017),但在昆虫中情况并不相同,所以GC/GC含量在SSR中的差异具体有什么作用,还需进一步验证。此外,有研究表明,SSR的多态性与其长度以及重复基序的重复次数相关,长度越长、重复基序的重复次数越多,其多态性越高(Megleczetal.,2012),在李白盾蚧转录组中,SSR位点的长度集中在10~15 bp中,占SSR总数的59.69%,而且还有28.15%长度>20 bp的SSR位点,由此推测李白盾蚧可能具有较高的遗传多态性,而这也暗示了该虫具有较强的入侵能力,能够较为轻松的在入侵地定殖,并进一步造成危害。
本研究中,基于转录组数据设计合成的引物多达12 538对,随机挑选50对引物后,首先利用宁夏银川的单头李白盾蚧DNA样本进行PCR扩增,共有34对引物成功扩增出与预期片段大小一致的特异性片段,后使用6个我国不同地理种群的李白盾蚧样本进行扩增稳定性验证,其中引物PP_4、PP_8、PP_31、PP_39在海南海口种群中无扩增条带,或条带模糊,表明这4对引物的扩增稳定性不高,在某些种群中不能成功扩增出目的条带,所以不能作为微卫星位点;引物PP_38在贵州雷山种群的扩增条带与目的条带大小不一致,说明存在非特异扩增,也不能作为微卫星位点。综上所述仍有29对引物可以稳定扩增,扩增效率达到了58%,说明本研究基于转录组数据预测的SSR位点数目可观,但其多态性还需进一步验证,以满足后续遗传多样性分析。
综上所述,本研究通过对李白盾蚧转录组数据进行分析以及对SSR位点的挖掘,结果表明,利用转录组数据开发微卫星位点高效可行,且为李白盾蚧之后的功能基因挖掘、种群遗传等研究奠定了基础。同时也有利于了解该虫的入侵机制、入侵来源以及在我国的扩散路线,对进一步综合防治该虫提供了理论依据,还可以对李白盾蚧的微卫星位点进行迁移,探索其在近缘物种桑白蚧中的扩增能力,判断这两种物种的亲缘关系。
