双组分调节系统对肺炎克雷伯菌多黏菌素耐药机制影响的研究进展
2023-11-09杨文丽王东亮冯钧帅齐保立史惠文
杨文丽,王东亮,冯钧帅,陈 莉,齐保立,史惠文,袁 媛
1. 甘肃中医药大学第一临床医学院(兰州 730000)
2. 甘肃省人民医院重症医学科(兰州 730000)
肺炎克雷伯菌(Klebsiella pneumoniae, KP)是一种重要的人类病原体,被归类为一种常见的机会性医院相关细菌,涉及多种感染,包括尿路感染、肺部感染、肝脓肿和手术伤口感染[1]。在过去十年中,全球范围内耐多药KP 分离株的报告显著增加。
多黏菌素(多黏菌素B 和黏菌素)是于1947年从多黏菌芽孢杆菌中分离出的一种具有抗革兰氏阴性菌活性的多阳离子抗菌肽[2]。在20 世纪80 年代,由于多黏菌素的神经毒性和肾毒性,其应用受到限制。21 世纪初,随着多重耐药(multi-drug resistance, MDR)微生物数量的增加,尤其是碳青霉烯耐药菌的出现,多黏菌素再次受到临床的关注[3]。多黏菌素是治疗KP 感染的最后手段药物,然而临床中出现了不同程度的耐碳青霉烯类肺炎克雷伯菌(carbapenem-resistantKlebsiella pneumoniae, CRKP)耐多黏菌素的情况。2022 年中国细菌耐药监测报告数据显示,CRKP 对多黏菌素B 的耐药率为7.9%,对黏菌素的耐药率为8.2%[4]。研究显示,双组分调节系统(two-component regulatory systems, TCSs)可通过感知环境变化调节KP 对多黏菌素耐药的修饰[5]。KP 可通过TCSs 修饰脂多糖(lipopolysaccharide,LPS)、mgrB 负反馈调节因子、外排泵等介导多黏菌素耐药。
1 细菌双组分调节信号转导的基础
典型的TCSs 由一对蛋白质组成,包括一个传感器组氨酸激酶(histidine kinase, HK)和一个反应调节因子(response regulator, RR)。可变HK n 端结构域感知环境变化,通过结合细胞外配体或通过其他构象变化,三磷酸腺苷(adenosine triphosphate, ATP)水解触发保守的c 端组氨酸残基的自磷酸化。HK 结合的磷酸被转移到RR 保守的n 端结构域的天冬氨酸残基上,RR 是一种位于细胞质中的同型二聚体蛋白,激活RR 的可变c 端输出结构域,允许其通过靶向DNA 来调节基因组表达。通过磷酸化的信息流,细菌能够有效感知其周围环境的变化(营养、pH 值、渗透压、抗生素等),并以一种允许对动态环境进行快速反应的方式协调基因表达[5]。
2 TCSs对多黏菌素耐药的影响机制
细菌细胞外膜(outer membrane, OM)的阴离子性质是由LPS 的存在所决定的,它包含一个带负电荷的脂质A 部分。LPS 是多黏菌素的初始靶点,多黏菌素可与LPS 的脂质A 区域的磷酸基相互作用[2,6]。一旦附着,多黏菌素就会通过取代二价阳离子Ca2+和Mg2+来破坏OM,导致细胞死亡。
当暴露在阳离子抗菌肽,如多黏菌素B和黏菌素下,细菌会通过改变LPS 保护自己免受不利环境的刺激。暴露在阳离子抗菌肽下激活了细菌的TCSs,从而导致磷酸乙醇胺(phosphoethanolamine, PEtN)和4-氨基阿拉伯糖(4-aminoarabinose, L-Ara4N)阳离子基团的合成和转移,对LPS 的脂质A 进行共价修饰[7]。这些LPS 共价修饰可以中和OM 上的负电荷,使OM 带正电荷,阻碍多黏菌素与OM 的结合,导致抗生素作用的减少或消失,从而形成多黏菌素耐药[8]。
3 PhoP/PhoQ和PmrA/PmrB对肺炎克雷伯菌的耐药机制
在介导KP 对多黏菌素耐药中最典型的两个TCSs 是PhoP/PhoQ 和PmrA/PmrB。研 究 发 现,在暴露于多黏菌素的KP 中,PhoP/PhoQ 和PmrA/PmrB 系统表达上调[9]。Naha 等[10]的研究显示,在多黏菌素耐药的KP 大多数菌株的TCSs 基因表达上调,包括PhoP(4~6 倍)、PhoQ(5~9 倍)、PmrA(4~13 倍)和PmrB(6~11 倍)。
PhoP/PhoQ 和PmrA/PmrB 的激活是由环境刺激和TCSs 内的特定突变触发的,这些突变导致它们的构成激活和随后的过表达[11-12]。
3.1 PhoP/PhoQ系统
PhoP/PhoQ 系统是KP 中研究最广泛的TCSs 系统,其在许多致病性和非致病性细菌中较为保守。
在各种致病菌中,PhoP/PhoQ 具有感知宿主细胞内信号并在感染期间调节细菌生活方式的能力[13]。反应调节因子PhoP 在许多革兰氏阴性菌中高度保守,传感器激酶PhoQ 位于细胞内膜(intracellular membrane, IM),可通过自磷酸化对几种环境变化作出反应,包括低pH 值、低Mg2+浓度和抗生素的存在,然后磷酸基团被转移到反应调节因子PhoP,它激活了参与LPS 修饰的下游基因的表达,通过遗传调控对OM 进行修饰[14-17]。
PhoP/PhoQ 系统的激活,主要受到mgrB基因突变的负反馈系统调节作用的影响,PhoP/PhoQ系统通过PmrD 间接激活PmrA/PmrB 系统来促进多黏菌素抗性[18-20]。mgrB的插入失活与PhoP/PhoQ 系统和pmrHFIJKLM 操纵子的过表达有关,PhoP/PhoQ 系统一旦被激活,磷酸化的PmrA 就会结合到pmrCAB 操纵子和pmrHFIJKLM 操纵子的启动子区域,增加RNA 聚合酶的识别和结合,并导致操纵子的上调,介导PEtN 与L-Ara4N 的合成和转移到脂质A[21]。
多黏菌素对易感表型抗性的逆转与PhoP和PhoQ基因的非同义突变相关。在智利的一项研究中,ST25 中PhoP 的104 位氨基酸替换、ST1161 中PhoQ 的A351D 氨基酸替换在黏菌素异质性耐药分离株中被检测到[22]。另外,PhoP基因的L26Q 突变可导致黏菌素耐药[23-24]。罗马尼亚的一项研究提示PhoP基因的L4F 突变、PhoQ基因的L26Q、Q426L、L224Q、Q317K 突变也会增加黏菌素的抗性[25]。此外,Halaby 等[26]在黏菌素异质性耐药(colistin heteroresistant, CST-HR)和产生超广谱β-内酰胺酶(extended-spectrum betalactamase, ESBL)的KP 分离物中发现了PhoQ 中的A21S 替代,使黏菌素最低抑制浓度(minimum inhibitory concentration, MIC) 从2 µg/mL 增 加 到16 µg/mL,表明该氨基酸替代在CST-HR 的表型中发挥了重要作用。
mgrB基因编码一个小的跨膜蛋白是PhoP/PhoQ 系统的负调控因子。mgrB的突变导致PhoP/PhoQ 双组分系统上调,进而导致pmrCAB和pmrHFIJKLM 操纵子的过表达[27-29]。pmrCAB和pmrHFIJKLM 操纵子的上调归因于磷酸化的PmrA,PmrA 被PmrD 激活,而PmrD 反过来又被mgrB破坏导致PhoP 磷酸化激活[9,30]。此外,激活的PhoP 也可直接激活KP 中的pmrHFIJKLM操 纵 子[31]。与TCSs 突 变(PmrA/PmrB 或PhoP/PhoQ)相比,mgrB突变/失活导致KP 对多黏菌素耐药的作用更大[24]。这一结果表明,mgrB在KP 的多黏菌素耐药性中起着重要作用。印度的一项研究发现,IS903 样和ISKpn26 样插入元件造成的mgrB的破坏导致KP 对黏菌素敏感性降低[32]。
有研究称IS5 样插入元件是KP 中mgrB破坏的主要元素,但ISKpn14(IS1 家族成员)的插入也偶有报道[33-34]。Morales-León 等[22]的研究发现IS5 样和IS1 样转座子插入序列使mgrB失活,降低了mgrB的mRNA 水平,从而影响多黏菌素的抗性。然而,沙特阿拉伯的一项研究发现存 在ISKpn14、ISKpn28、IS903 导 致mgrB基 因破坏,而ISKpn14 是主要的IS,59%的分离株存在mgrB基因破坏是由ISKpn14 介导的[23]。在罗马尼亚的一项研究中,ISL3(ISKpn25)和IS5(ISKpn26)在不同的核苷酸位置破坏mgrB基因,增加CPKP 的黏菌素抗性[25]。另外,生物信息学工具预测mgrB基因M27K 突变,提示同样是有害的。相比之下,Liu 等[35]研究发现,MgrB 蛋白的表达不受M27K 突变的影响。
在Naha 等[10]的研究中,mgrB中的氨基酸替换(C28G,V32G)导致突变是有害的,影响主要功能域,可能调节蛋白活性,有助于黏菌素抗性的增加。mgrB的失活也被证明可以通过抑制宿主防御反应的启动和限制多种抗菌肽的作用来增强KP 分离株的毒力[36]。
3.2 PmrA/PmrB系统
KP 的多黏菌素耐药也归因于PmrA/PmrB基因的突变,该基因是另一个编码控制L-Ara4N 和PEtN 合成的双组分调节系统[22,37-38]。
在大肠杆菌和肠道沙门氏菌中,PmrB 作为一种传感器细胞质膜结合激酶,被高价铁(Fe3+)和酸性环境(pH=5.5)激活。激活后,它通过磷酸化激活PmrA[39]。在体内,PmrA/PmrB 参与感知环境,帮助细菌在体内侵袭上皮细胞、在细胞内生长、细胞间扩散、诱导巨噬细胞凋亡,并且与细菌毒力表达相关[40]。PmrA或PmrB基因的突变通常会导致PmrA 的结构性激活,可以上调pmrCAB 操纵子和pmrHFIJKLM 操纵子,pmrCAB操纵子参与PEtN 对脂质A 修饰,而pmrHFIJKLM操纵子编码的酶负责合成和将L-Ara4N 转移到脂质A[40]。总的来说,PmrA 的这种构成性激活导致了更多正电荷的LPS,从而降低了带正电荷的多黏菌素的亲和力。
PmrB基因的突变已在具有耐药性或敏感性降低的KP 分离株中被广泛描述。一项研究显示,CRKP 中PmrB基因P95L、T157P、R256G、V352E 突变与黏菌素的耐药性增加相关[25]。此外,Jayol 等[41]描述了6 个黏菌素耐药菌株的PmrB 中一个单一的T157P 氨基酸取代,而这种取代会破坏PmrB 蛋白的α-螺旋二级结构,并连续激活PmrA,从而产生PmrC 的过表达,最终导致黏菌素抗性。另一项研究在ST25 和ST11 中也发现了PmrB 的相关氨基酸(分别是P95L 氨基酸和A256G 氨基酸)取代导致KP 对黏菌素的耐药性增加[22]。而Liu 等[35]的研究显示,PmrB 中的氨基取代D313N 使黏菌素MIC 提高到8 mg/L,比原菌株高16 倍,而PmrB 中的M285L 和PmrA 中的S204L 并没有增加黏菌素对原菌株的MIC 值。
Wand 等[42]的研究在PmrA中只发现了G53C突变导致黏菌素MIC 增加到64 mg/L。目前,关于PmrA基因突变导致多黏菌素耐药性增加的研究相对较少。
PmrA/PmrB 和PhoP/PhoQ 的突变和构成性激活可能共同导致pmrCAB 操纵子和pmrHFIJKLM操纵子的激活,从而导致多黏菌素耐药性[2,8]。与多黏菌素抗性相关的PhoP/PhoQ 和PmrA/PmrB 调控关系见图1。
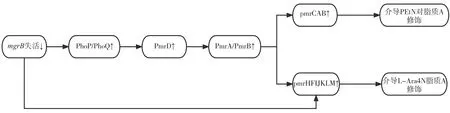
图1 与多黏菌素抗性相关的PhoP/PhoQ和PmrA/PmrB调控图Figure 1. Regulatory plots of PhoP/PhoQ and PmrA/PmrB associated with polymyxin resistance
4 其它TCSs对肺炎克雷伯菌的耐药机制
进化实验表明,KP 中多黏菌素耐药性也可能由于CrrB基因的突变而出现[43]。CrrA/CrrB 双组分系统通过PmrAB 调控网络参与了对pmrHFIJKLM 和CrrC 操纵子的控制,而CrrB 调控pmrHFIJKLM 操纵子的表达是通过CrrC 操纵子介导的[44]。研究发现,临床CrrB 突变导致L-Ara4N和PEtN 同时添加到脂质A 中,诱导更高的多黏菌素抗性[45]。Naha 等[10]的研究也提示,CrrA/CrrB突变导致KP 对黏菌素产生耐药性。在罗马尼亚的一项研究中,CrrB基因罕见的P151S 替换,使异质性耐药KP 的黏菌素MIC >64 mg/L[25]。另一项研究已证实GrrB基因突变可诱导KP 对黏菌素的耐药性升高,且该研究发现CrrB基因Q10L、Y31H、W140R、N141I 和S195N 错义突变均会引起KP 对黏菌素的MIC 值增加64~1024 倍[44]。类似地,Jayol 等[46]报道了一个具有细微差异的突变,CrrB中的P151L 赋予了黏菌素耐药性,而Pitt 等[47]证实了同一基因中的P158R 替换。此外,CrrA/CrrB 可激活PmrA,而CrrB的功能获得突变可激活导致黏菌素耐药性的基因表达,而不受PmrA/PmrB 系统的影响[48-49]。
KP 编码另外两种TCSs 为CpxAR 和PhoBR,它们抑制KpnO 孔蛋白,降低了对多种抗生素的敏感性,包括氯霉素、阿米卡星、萘啶酸和四环素[50-51]。两个TCSs 都由于OM 在应激状态下而激活。此外,在KP 中CpxA/CpxR 可同时上调三个MDR RND 家族外排泵以响应OM 应激[50]。
TCSs 对多种细菌的生存发挥着关键作用,它为细菌提供了感知和反应周围环境的不可或缺的工具。鉴于TCSs 在细菌稳态中的重要作用以及可通过它们激活的多种抗生素耐药性机制,TCSs是开发新的抗菌疗法的一个较好靶点[52-53]。使用预测软件来识别结合一个或多个TCSs 以使其无效的假定化合物的能力,将是药物靶向TCSs 的一个重要工具[54]。例如PmrB基因,71%的耐药性非同义突变发生在Hamp(存在于组氨酸激酶、腺苷酸环化酶、甲基接受蛋白和磷酸酶)连接子和DHp(二聚化和组氨酸磷酸转移)结构域。这些结果增强了研究人员对支持多黏菌素耐药性的调控机制的理解,并可能有助于开发新的策略来减少耐药性的出现[55]。此外,有研究显示,与对碳青霉烯类抗生素敏感的菌株相比,CRKP 中OmpK35/36 孔蛋白减少与EnvZ-OmpR、PhoPQ 和BaeSR 等TCSs 的下调有关[56]。
5 结语
耐多药KP 是一项全球性的公共卫生问题。本文通过阐述TCSs 导致KP 对多黏菌素耐药的机制,将TCSs 设想为未来可能的抗菌靶点,为规划抗击KP 耐药性的策略提供依据,拓宽耐药菌研究的视野,从而为未来的研究提供更多思路,并为临床上更加有效的治疗耐多药KP 感染提供参考。
