UPLC-MS/MS测定大黄蛰虫丸中的9种成分
2023-11-07王建成韩庆霞
刘 丽,王建成,韩庆霞,王 亮,王 娜
(临沂市检验检测中心,山东 临沂 276001)
大黄蛰虫丸源于汉代医圣张仲景编著的《金匮要略》,收载于《中国药典》2020年版一部[1],主要由熟大黄、干漆(煅)、桃仁、炒苦杏仁、黄芩、地黄、白芍、甘草、土鳖虫(炒)、水蛭(制)、虻虫(去翅足,炒)、蛴螬(炒)等十二味中药组成,同时含有植物药和动物药;具有活血破瘀、通经消癥的功能,用于瘀血内停所致的癥瘕、闭经,症见腹部肿块、肌肤甲错、面色黯黑、潮热羸瘦、经闭不行[1],可用于慢性肝炎、肝纤维化和肿瘤的辅助治疗等[2-9]。《中国药典》2020年版一部中对大黄蛰虫丸含量的测定仅包括大黄素和大黄酚[1],缺乏对其他有效成分含量的控制。大黄蛰虫丸药效需要复方协同发挥作用,多成分全面质量控制具有重要意义,由于其他动物药目前缺乏明晰的有效成分指标,因此,本文参考《中国药典》中桃仁、苦杏仁、黄芩、白芍等质量控制指标,选取黄芩素、黄芩苷、苦杏仁苷、大黄酚、大黄素甲醚、甘草苷、芍药苷、毛蕊花糖苷、甘草酸铵等9种成分的含量对大黄蛰虫丸进行质量控制。
目前,已报道的大黄蛰虫丸成分测定主要方法有高效液相色谱法(HPLC)[1,10-13],但多种成分同时测定的方法较少。HPLC在测定中需要对每个化合物的最大吸收波长进行扫描获取,同时在多组分测定中需要切换波长;与质谱检测器相比,紫外检测器灵敏度较低,对液相色谱分离技术也提出更高要求。采用超高效液相色谱-质谱联用技术(UPLC-MS/MS),以液相色谱作为分离系统,质谱为检测系统,样品在质谱部分和流动相分离,被离子化后,经质谱的质量分析器将离子碎片按质量数分开,经检测器得到质谱图;具有高灵敏度、对色谱分离要求相对简单、多成分同时测定、检测效率高等优点。为了保证测定的灵敏度,UPLC-MS/MS只需特定的离子对即可[14]。UPLC-MS/MS广泛用于生化分析、药代动力学、食品安全、化妆品和保健食品分析和环境污染物分析等领域[15-22]。本文采用UPLC-MS/MS,通过多反应监测模式(MRM)和正负离子同时采集方式,在一个分析周期内完成对样品溶液中黄芩素、黄芩苷、苦杏仁苷、大黄酚、大黄素甲醚、甘草苷、芍药苷、毛蕊花糖苷、甘草酸铵等9种成分的扫描,得到准确质量数和准确碎片离子信息。
1 仪器与试药
1.1 仪器
Exion LC AD/Triple Quad 4500超高效液相色谱-质谱联用仪(美国SCIEX公司),配备有ESI源,Exion LC AD超高效液相色谱仪配备高压二元泵(最大耐压100 MPa)、自动进样器(温控)、柱温箱(温控)、在线控制器。Analyst Software 3.2软件,用于仪器控制、设置化合物参数、数据采集。SCIEX OS软件用于数据处理。KQ-300 GDV温控超声仪(昆山市超声仪器公司)。Mettler XP 205电子天平(梅特勒-托利多公司)。
1.2 试药
黄芩素对照品(批号:111595-201808,含量以97.9 %计)、黄芩苷对照品(批号:110715-201821,含量以95.4 %计)、苦杏仁苷对照品(批号:110820-201808,含量以88.2 %计)、大黄酚对照品(批号:110796-201922,含量以99.4 %计)、大黄素甲醚对照品(批号:110758-201817,含量以99.2 %计)、甘草苷对照品(批号:111610-201908,含量以95.0 %计)、芍药苷对照品(批号:110736-202044,含量以96.8 %计)、毛蕊花糖苷对照品(批号:111530-201914,含量以95.2 %计)、甘草酸铵对照品(批号:110731-202021,含量以96.2 %计),均购自中国食品药品检定研究院。
乙腈为色谱纯(美国默克试剂公司);甲酸为色谱纯(上海阿拉丁);水为超纯水(Milipore超纯水机);其他试剂均为分析纯。5批不同厂家生产大黄蛰虫丸(市售)。
2 方法与结果
2.1 液相色谱-质谱联用条件
2.1.1 色谱条件 色谱柱:Agilent Technologies Infinity Lab Poroshell 120 EC-C18(4.6 mm×100 mm,2.7 μm);流动相:乙腈(A)-0.1 %甲酸水溶液(B)进行梯度洗脱(0~2 min,70 % B;2~12 min,10 % B;12~15 min,70 % B);自动进样器温度:15 ℃;柱温:40 ℃;流速:0.3 ml/min;进样量:5 μl。
2.1.2 质谱条件 Triple Quad 4500质谱系统配有Turbo V ESI源,采用MRM和正负离子同时扫描模式,电子喷雾电压为+5500 V/-4500 V,离子源温度为500 ℃,气帘气使用氮气,流速为25.0 psi,碰撞气使用氮气,流速为9.0 psi,喷雾气使用空气,流速为55.0 psi,辅助加热气为空气,流速为55.0 psi。使用SCIEX质谱专用校正液进行PPG质量准确度校准一次质量轴。使用Q1 Scan方式确定化合物母离子的质荷比,准确到小数点后一位,母离子扫描范围为50.0~1300.0 m/z,针泵恒流进样流速设为10.000 μl/min,持续时间(Duration)设为5.00 min,正负模式分开扫描。使用Product Ion Scan(MS2)确定子离子的质荷比,准确到小数点后一位,Product Of设为母离子质荷比,定持续时间(Duration)为2.00 min,设定碰撞电压(CE)初始值为5 eV,可按需求以5 eV为步长手动调节,母离子的强度为图谱中基峰强度的1/3到1/4为宜,待仪器稳定后选择至少2~3个子离子。使用MRM优化化合物参数,驻留时间(Dwell Time)设为100 ms,用“Ramp”优化碰撞能量(Collision Energy)和去簇电压(Declustering Potential),连续优化2次,以得到较确切结果。黄芩素、黄芩苷、苦杏仁苷、大黄酚、大黄素甲醚、甘草苷、芍药苷、毛蕊花糖苷、甘草酸铵等9种化合物的保留时间、准确质量数、二级碎片离子信息、去簇电压、碰撞能量见表1。混合对照品溶液和供试品溶液总离子流图谱见图1。
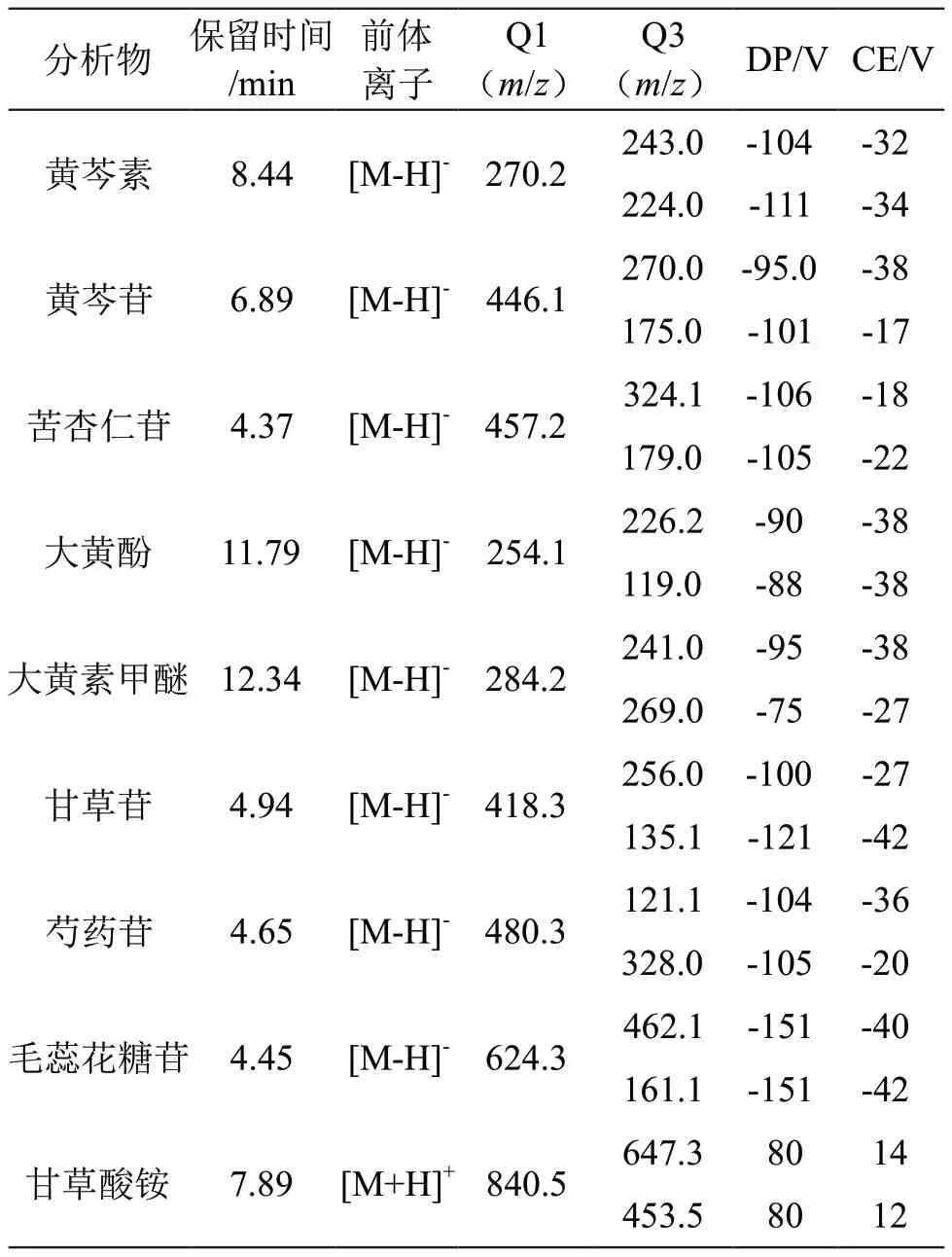
表1 9种成分的保留时间和质谱参数

图1 混合对照品溶液(A)和供试品溶液(B)的总离子流图
2.2 溶液配制
2.2.1 系列浓度混合对照品溶液 分别精密称取黄芩素、黄芩苷、苦杏仁苷、大黄酚、大黄素甲醚、甘草苷、芍药苷、毛蕊花糖苷、甘草酸铵对照品各约10 mg,置入50 ml量瓶,加甲醇溶解并稀释至刻度,摇匀,作为混合对照品贮备液(约0.2 mg/ml);再分别精密吸取上述贮备液适量,加甲醇溶液逐步稀释得到0.04,0.08,0.10,0.20,0.40,0.80,1.0 μg/ml的系列浓度混合对照品溶液。
2.2.2 供试品溶液 取供试品,研细后,精密称取细粉约3.0 g,置入50 ml量瓶,加甲醇约30 ml,超声30 min(功率160 W,频率40 kHz),放冷,加甲醇稀释至刻度,摇匀,0.22 μm微孔滤膜滤过。精密量取续滤液10 ml,置入100 ml量瓶,用10 %甲醇-水溶液稀释至刻度,摇匀,即得。
2.3 方法学考察
2.3.1 专属性考察 取对照品、供试品溶液适量,按2.1项条件测定,结果见图1。由图1可见,各成分离子对和溶剂对测定无干扰,表明该方法专属性良好。
2.3.2 线性关系考察 分别精密吸取系列混合对照品溶液各5 μl,注入液相色谱仪,记录峰面积;以待测成分质量浓度X为横坐标,峰面积Y为纵坐标绘制标准曲线并进行线性回归,黄芩素、黄芩苷、苦杏仁苷、大黄酚、大黄素甲醚、甘草苷、芍药苷、毛蕊花糖苷、甘草酸铵等9种成分的线性范围、回归方程、相关系数见表2。结果表明,各成分在各自范围内线性关系良好。
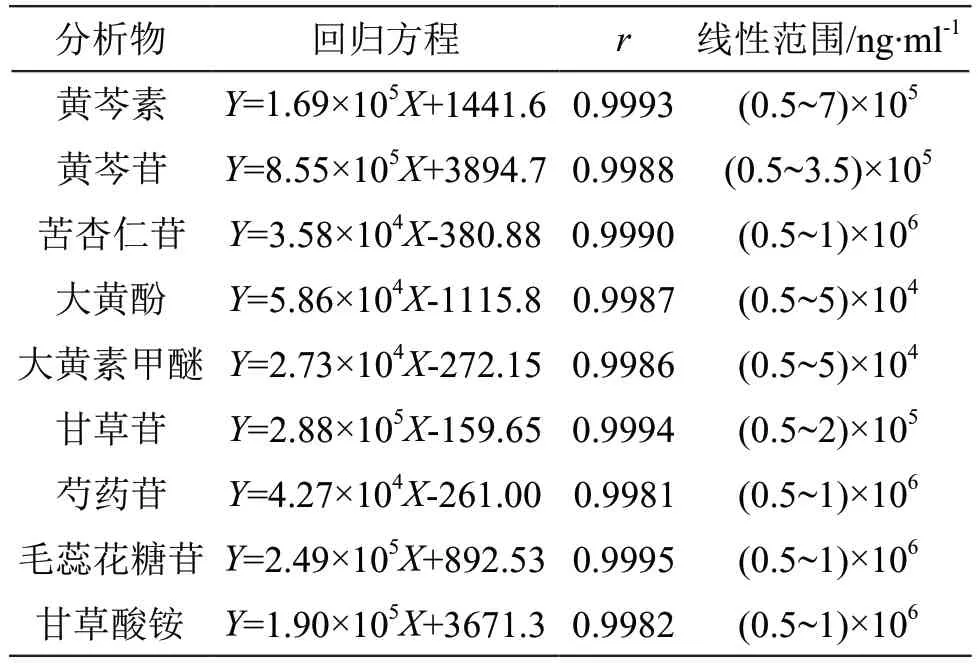
表2 9种成分的回归方程、相关系数、线性范围
2.3.3 检测限和定量限 将混合对照品溶液稀释至不同浓度,进样测定,以信噪比3:1为检出限和10:1为定量限,计算得黄芩素、黄芩苷、苦杏仁苷、大黄酚、大黄素甲醚、甘草苷、芍药苷、毛蕊花糖苷、甘草酸铵等9种成分的检出限分别为0.1,0.1,0.2,0.1,0.1,0.2,0.1,0.2,0.2 ng/ml,定量限均为0.5 ng/ml。
2.3.4 精密度试验 取同一混合对照品溶液0.2 μg/ml续进样6次,记录峰面积,计算得黄芩素、黄芩苷、苦杏仁苷、大黄酚、大黄素甲醚、甘草苷、芍药苷、毛蕊花糖苷、甘草酸铵等9种成分的峰面积的RSD分别为1.53 %,1.27 %,1.60 %,1.09 %,1.46 %,1.38 %,1.17 %,1.84 %,1.03 %,均小于2.0 %,表明精密度良好。
2.3.5 重复性试验 分别取同一批号大黄蛰虫丸样品6份,精密称定,按2.2.2项下方法制备供试品溶液,按2.1项下色谱条件分别进样,记录峰面积,分别计算得黄芩素、黄芩苷、苦杏仁苷、大黄酚、大黄素甲醚、甘草苷、芍药苷、毛蕊花糖苷、甘草酸铵在6份样品中的含量的RSD分别为1.37 %,1.62 %,1.24 %,1.68 %,1.25 %,2.02 %,1.63 %,1.78 %,2.11 %,表明本方法的重复性良好。
2.3.6 稳定性试验 取2.3.5项下同一供试品溶液(4 ℃冰箱中储存备用),分别于0,2,4,8,16,24,48 h分别按2.1项下色谱条件进样,记录峰面积,计算得黄芩素、黄芩苷、苦杏仁苷、大黄酚、大黄素甲醚、甘草苷、芍药苷、毛蕊花糖苷、甘草酸铵等9种成分的峰面积的RSD分别为1.52 %,1.14 %,1.73 %,1.01 %,1.62 %,1.84 %,1.33 %,2.20 %,1.42 %,均小于3.0 %,表明供试品溶液在48 h内稳定性良好。取同一混合对照品溶液(4 ℃冰箱中储存备用),分别于0,2,4,8,16,24,48 h分别按2.1项下色谱条件进样,记录峰面积,计算得黄芩素、黄芩苷、苦杏仁苷、大黄酚、大黄素甲醚、甘草苷、芍药苷、毛蕊花糖苷、甘草酸铵等9种成分峰面积的RSD分别为1.37 %,1.08 %,1.55 %,1.02 %,1.57 %,1.68 %,1.21 %,1.89 %,1.33 %,均小于3.0 %,表明对照品溶液在48 h内稳定性良好。
2.3.7 回收率试验 取黄芩素、黄芩苷、苦杏仁苷、大黄酚、大黄素甲醚、甘草苷、芍药苷、毛蕊花糖苷、甘草酸铵对照品适量,精密称定,加甲醇制成每1 ml含黄芩素75 μg、黄芩苷135 μg、苦杏仁苷170 μg、大黄酚400 μg、大黄素甲醚300 μg、甘草苷35 μg、芍药苷200 μg、毛蕊花糖苷30 μg、甘草酸铵20 μg的混合溶液,作为待加混合对照品溶液。取已测定黄芩素、黄芩苷、苦杏仁苷、大黄酚、大黄素甲醚、甘草苷、芍药苷、毛蕊花糖苷、甘草酸铵等9种成分含量的样品6份,各约1.5 g,精密称定,分别精密加入上述待加混合对照品溶液1 ml,按2.2.2项下方法制备供试品溶液。按2.1项下色谱条件分别进样测定,记录峰面积,计算回收率和RSD,结果见表3。
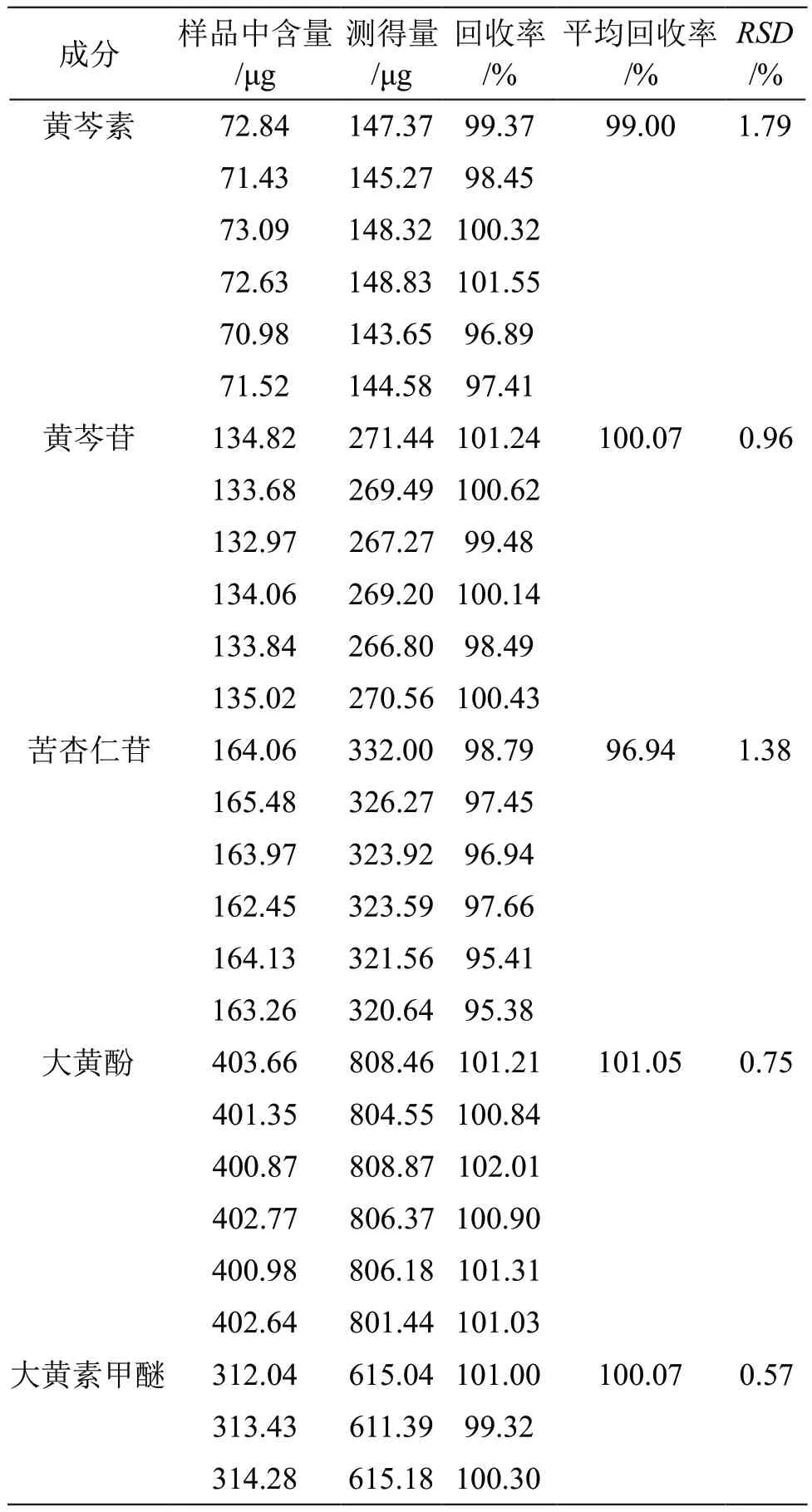
表3 回收率试验结果
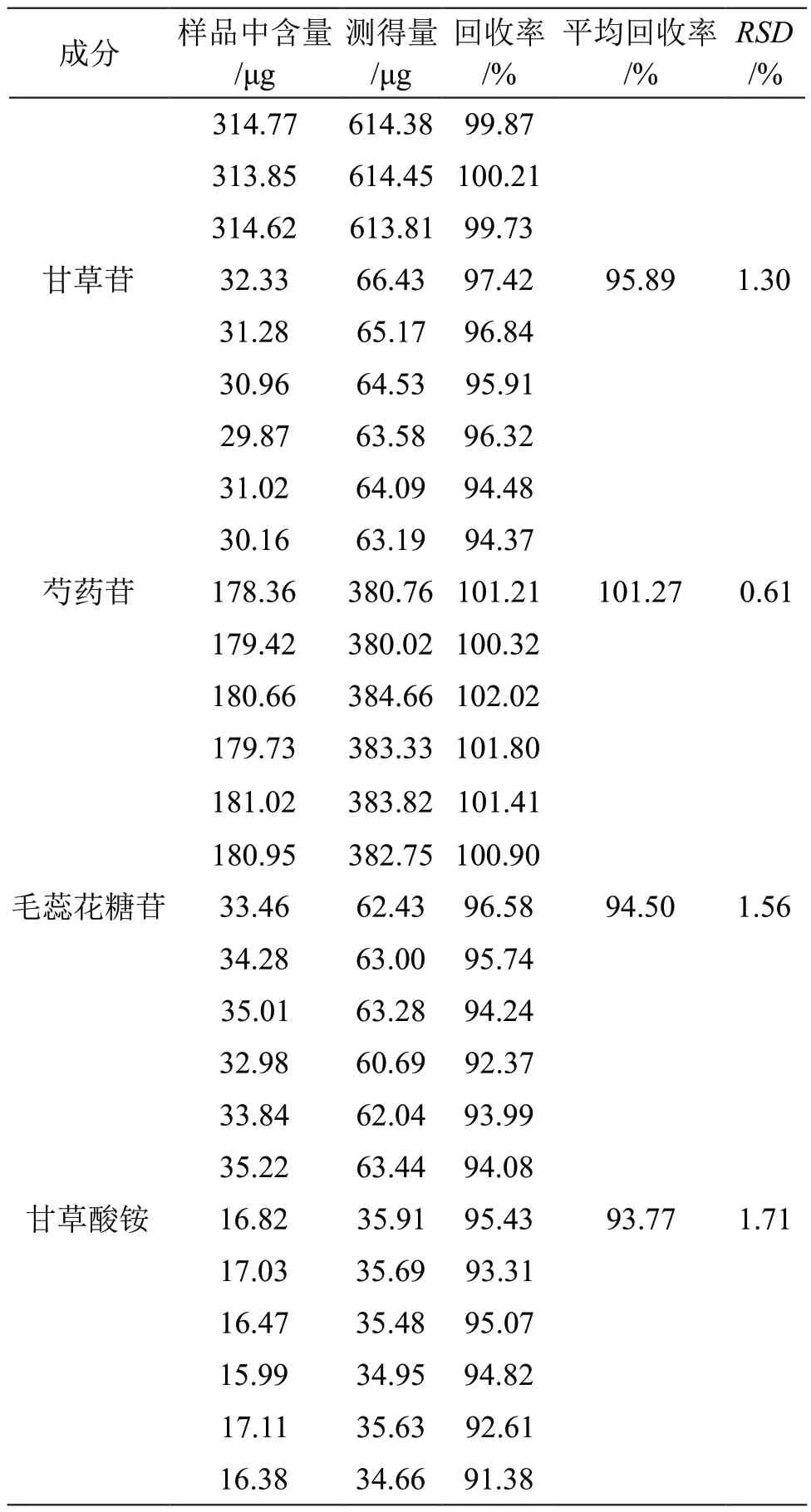
表3 (续)
2.4 样品测定
取5批大黄蛰虫丸样品,分别按2.2.2项下方法制备供试品溶液,按2.1项下色谱条件进样测定,记录峰面积,根据标准曲线计算含量,结果见表4。
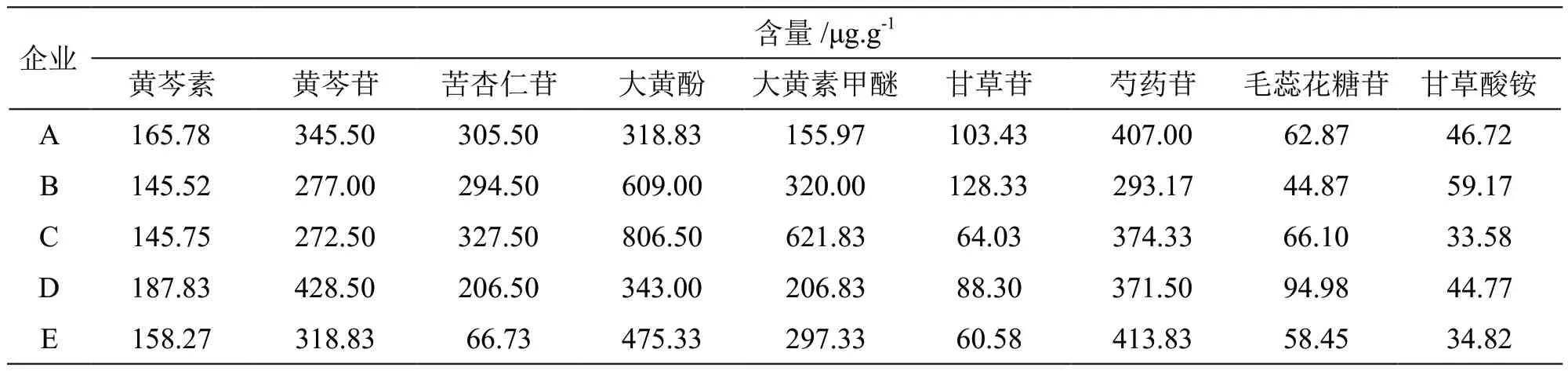
表4 9种成分的含量测定结果
3 讨论
3.1 检测方法的选择
大黄蛰虫丸处方中含12味药材,所含中药味较多,采用HPLC法检测时色谱峰干扰较多,方法灵敏度低,不能同时满足定性定量的需求。UPLCMS/MS具有高效快速的分离能力、超高的灵敏度和准确度、高选择性的特点,可在一针15 min内同时完成黄芩素、黄芩苷、大黄素甲醚、大黄酚、芍药苷、苦杏仁苷、毛蕊花糖苷、甘草苷、甘草酸铵等9种成分的定量测定和定性分析。
3.2 提取方法和提取溶剂的选择
本试验考察了超声提取、回流提取、甲醇-水(2:8)、甲醇-水(1:1)、乙腈-水(1:1)、乙腈、甲醇溶液的提取效果。结果表明,提取效率、峰的分离度和峰形差别明显,超声提取和甲醇提取的效果最好,因此选用超声提取和甲醇作为提取溶剂。测定时纯甲醇溶剂对峰形有影响,加入水溶液后,峰形和响应值得到明显改善。因此,最终确定用甲醇提取,并用10 %甲醇-水溶液稀释定容。
3.3 质谱扫描模式的选择
本实验采用MRM扫描模式,采用正负离子同时采集的方式,在一个分析周期内完成对样品溶液中黄芩素、黄芩苷、苦杏仁苷、大黄酚、大黄素甲醚、甘草苷、芍药苷、毛蕊花糖苷、甘草酸铵等9种成分的扫描,得到准确质量数和准确碎片离子信息,在完成定量分析的基础上还能同时得到化合物二级准确子离子信息,可作为定性分析的依据。
3.4 测定结果分析
5批样品中黄芩素、黄芩苷、苦杏仁苷、大黄酚、大黄素甲醚、甘草苷、芍药苷、毛蕊花糖苷、甘草酸铵等9种成分均有检出,但含量差别显著,目前,中成药的质量标准多采用控制某一成分的含量,并不能完全完整反映中成药的有效成分状况。因此,应尽量对多种有效成分同时测定,获得中药有效成分多维信息,使不同的企业间、同一企业的不同批次间的产品质量稳定,有效准确控制中药质量,确保疗效的稳定、可控。
