高效液相色谱-串联质谱法同时测定水产品中10 种硝基咪唑类药物残留量
2023-11-07谢云波童文羽朱志强石义付
谢云波,易 鸣,童文羽,曾 智,朱志强,石义付
(湖北省水产科学研究所,武汉 430071)
硝基咪唑(NMZs)类药物是一类合成的人和动物常用的广谱抗菌和抗原虫药物,在水产养殖中常被用于防治和治疗寄生虫感染[1],常见的有甲硝唑、洛硝哒唑和地美硝唑等。但由于硝基咪唑类药物含有的硝基杂环具有细胞诱变性、致癌性和潜在的致畸性,经生物体代谢后产生的带有侧链羟甲基化合物具有与原药相似的毒性,其残留对水产品质量安全构成了直接威胁[2],许多国家和地区已禁止该类药物用于食源性动物,中国也于2002 年将该类药物列入《食品动物禁用的兽药及其它化合物清单(中华人民共和国农业部公告第193 号)》[3]。
目前水产品中硝基咪唑类药物残留量检测的国家标准包括GB/T 21318—2007《动物源性食品中硝基咪唑残留量检测方法》[4]和《动物源性食品中4 种硝基咪唑残留检测液相色谱-串联质谱法(中华人民共和国农业部公告第1025 号)》[5],地方标准包括浙江省质量技术监督局发布的DB33/T 693—2008《动物源性食品中硝基咪唑类药物残留量的测定高效液相色谱法》[6],其中《动物源性食品中4 种硝基咪唑残留检测液相色谱-串联质谱法(中华人民共和国农业部公告第1025 号)》[5]仅能检测甲硝唑、羟甲基甲硝咪唑、洛硝哒唑、二甲硝唑;DB33/T 693—2008《动物源性食品中硝基咪唑类药物残留量的测定高效液相色谱法》[6]也只能检测甲硝唑、洛硝哒唑、地美硝唑、替硝唑、奥硝唑,且因为是液相法,灵敏度较低;GB/T 21318—2007《动物源性食品中硝基咪唑残留量检测方法》[4]能检测甲硝唑、洛硝哒唑、地美硝唑、氯甲硝咪唑、苯硝咪唑、异丙硝唑、2-甲硝咪唑、4-甲硝咪唑8 种原药和羟基甲硝唑、羟甲基甲硝咪唑2 种代谢产物,但该方法前处理操作繁琐,对实验室硬件和操作人员水平要求较高。而且以上国家和地方标准都是针对动物源性基质,没有针对水产品基质进行优化,实际试验过程中出现了标准曲线线性不佳,回收率不稳定的情况。
针对以上方法的不足,本研究建立了同时测定水产品中10 种硝基咪唑类药物(表1)含量的高效液相色谱串联质谱法,方法灵敏度可达1 μg/kg,线性范围为1~150 μg/L,适用于草鱼、加州鲈鱼、克氏原螯虾,该方法步骤简单、灵敏度高,适用于大批量样品的检测。
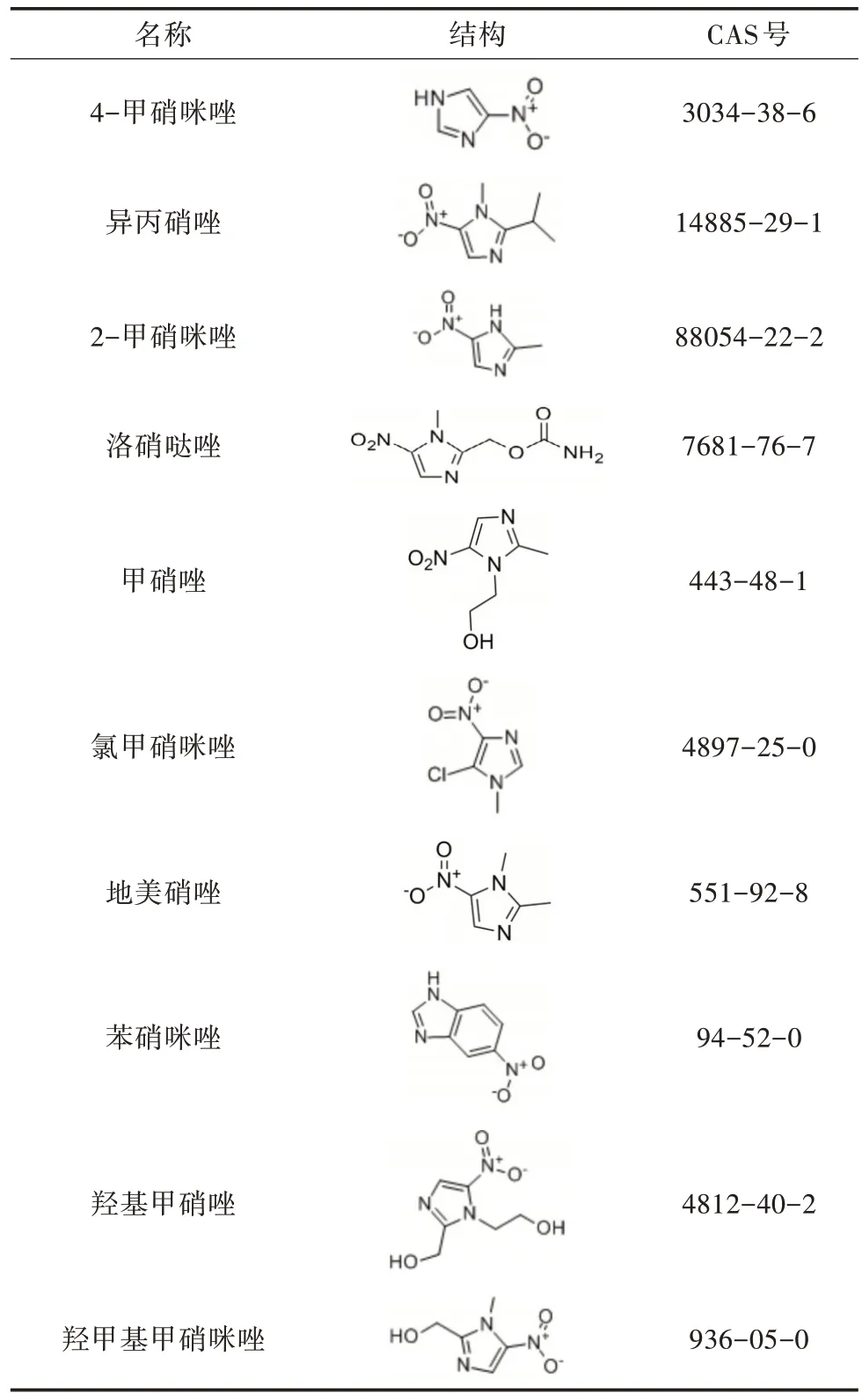
表1 10 种硝基咪唑类药物的结构和CAS 号
1 材料与方法
1.1 材料与试剂
试验样品均来自湖北省白沙洲农产品大市场,品种有草鱼、加州鲈鱼、克氏原螯虾。
选用10 种硝基咪唑类药物标准品,其生产批号、纯度、不确定度如表2 所示。
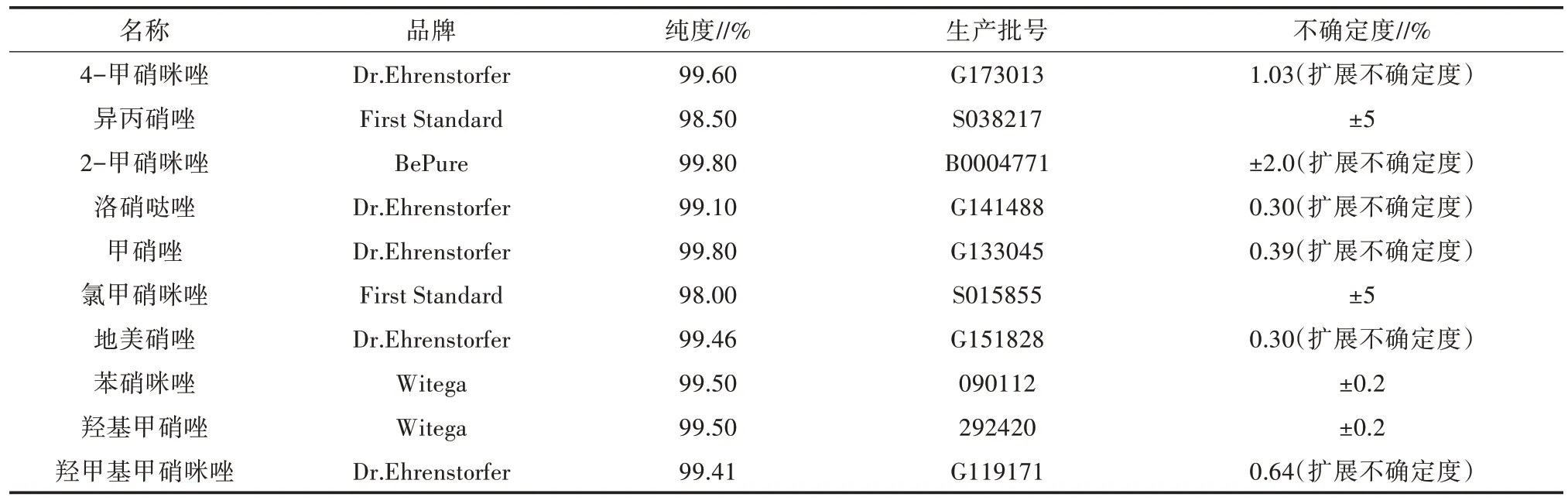
表2 硝基咪唑类药物标准品信息
乙酸乙酯、氨水、正己烷均为分析纯,购自国药集团化学试剂上海有限公司;甲醇、甲酸均为质谱纯,购自美国默克公司。
1.2 仪器
XPE205 型十万分之一天平(瑞士METTLER TOLEDO 公司);RE52CS 型旋转蒸发仪(上海亚荣生化仪器厂);KQ-500B 型超声波仪(昆山市超声仪器有限公司);U300 型超高效液相色谱串联SCIEX QTRAP 4500 质谱仪(美国赛默飞公司)。
1.3 方法
1.3.1 标准溶液配制 分别称取10 种硝基咪唑类药物标准品10 mg,用少量甲醇溶解,超声波助溶,置于100 mL 棕色容量瓶中定容,配制成100 mg/L 的标准储备液,4 ℃冷藏保存,有效期3 个月。分别量取10 种硝基咪唑类药物标准溶液,用甲醇稀释成浓度为0.5 mg/L 的混合标准工作液,现配现用。
1.3.2 样品制备 鱼去鳞,沿背脊取可食肌肉部分,虾去头、壳,取可食肌肉部分,切为小块,绞成肉糜,经GB/T 21318—2007《动物源性食品中硝基咪唑残留量检测方法》[4]液质法检测确认阴性,待用。
1.3.3 样品提取 称取5.00 g 样品,置于50 mL 离心管中,加入15 mL 乙酸乙酯,100 μL 氨水,加盖后垂直振荡(500 次/min)1 min,4 000 r/min 离心5 min,分离后取上清液,重复上述操作1 次,合并2 次上清液,40 ℃旋转减压蒸馏至干。
1.3.4 样品净化 向茄形瓶中加入2 mL 正己烷,涡旋1 min,再加1.5 mL 10%甲醇溶液,涡旋1 min,再将全部液体转移至离心管中,4 000 r/min 离心10 s,去除上层正己烷,吸取下层溶液,过0.22 μm 微孔滤膜待上机。
1.3.5 色谱条件 进样量:10 μL;色谱柱:岛津C18(150 mm×2 mm,5 μm);柱温:35 ℃。流动相A:0.2%甲酸水溶液;流动相B:乙腈;流速:0.4 mL/min,各组分梯度洗脱程序如表3 所示,同时为了减少离子源污染,设定进样开始后3.5~7.0 min 流动相才进入检测器。
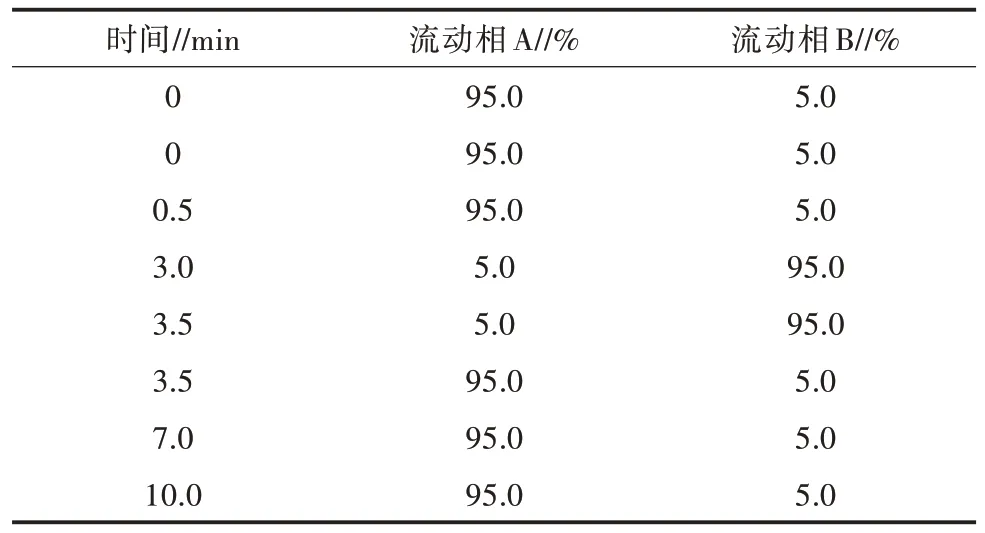
表3 洗脱程序
1.3.6 仪器质谱条件 离子源:ESI;扫描方式:正离子模式;检测方式:质谱多反应监测(MRM);电喷雾电压:5 500 V;雾化气压力:0.379 MPa;气帘气压力:0.207 MPa;辅助气压力:0.379 MPa;离子源温度:550 ℃,10 种硝基咪唑药物的保留时间、定量离子对、定性离子对、碰撞能量等见表4。
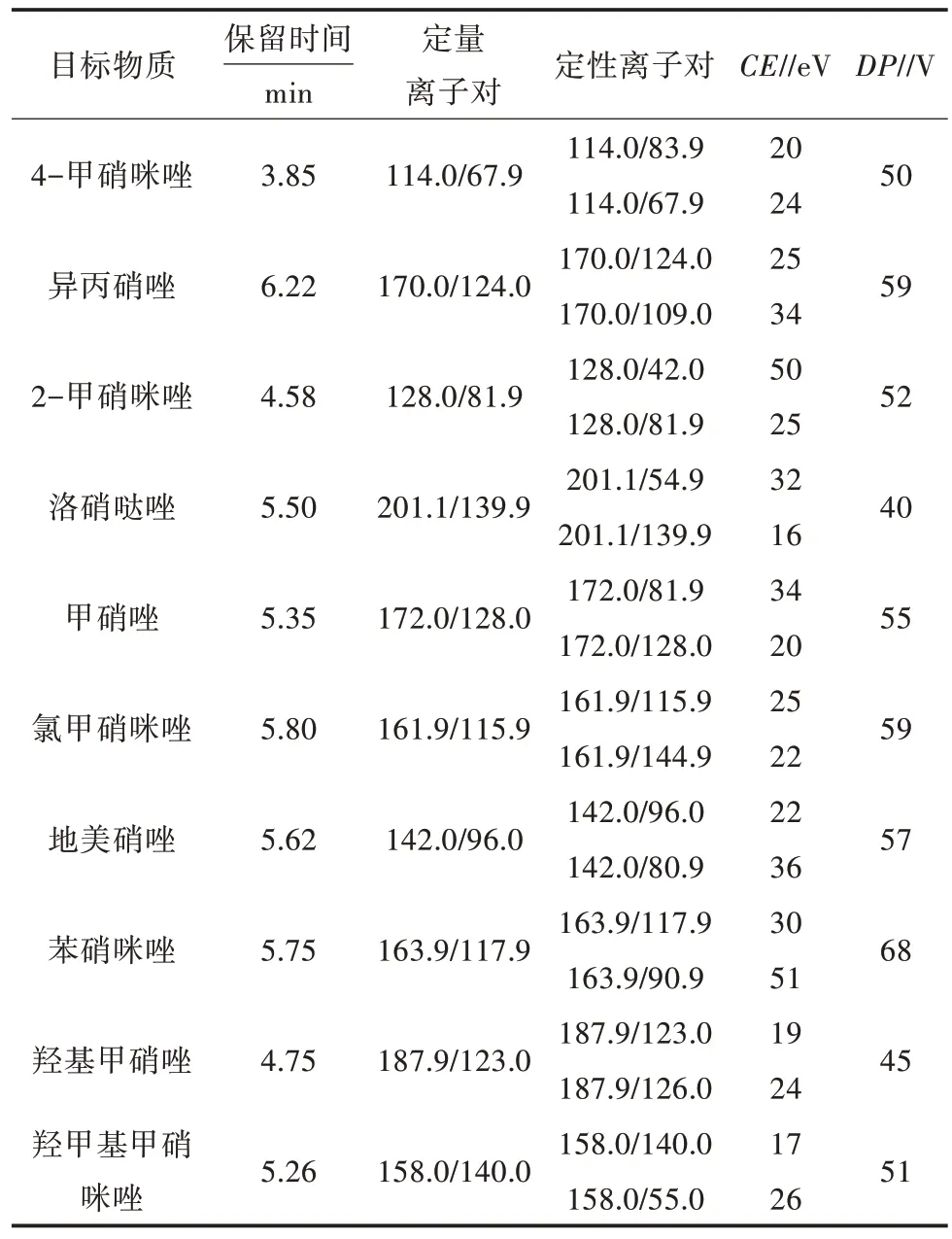
表4 10 种硝基咪唑药物的保留时间、选择监测离子设定参数
2 结果与分析
2.1 前处理优化
水产品中硝基咪唑类药物残留量的检测方法主要使用乙酸乙酯、甲醇、乙腈、二氯甲烷等有机溶剂作为提取剂,如《动物源性食品中4 种硝基咪唑残留检测液相色谱-串联质谱法(中华人民共和国农业部公告第1025 号)》[5]使用乙酸乙酯提取,正己烷液液萃取,MCX 固相萃取小柱净化,HPLC-MS 检测,外标法定量;方力等[7]用乙酸乙酯提取,经正己烷脱脂、50 mg 乙二胺-N-丙基硅烷(PSA)吸附剂吸附净化,经0.22 μm 亲水聚四氟乙烯(PTFE)滤膜过滤后直接进行上机检测;刘艳萍[8]和王志杰等[9]使用无水硫酸钠作为助剂,乙酸乙酯提取,HPLC-MS 检测,内标法定量;GB/T 21318—2007《动物源性食品中硝基咪唑残留量检测方法》[4]使用甲醇-丙酮均质或超声波提取,乙酸乙酯液液萃取,凝胶渗透色谱净化,再经C18 固相萃取小柱净化,HPLC-MS 检测,外标法定量;DB33/T 693—2008《动物源性食品中硝基咪唑类药物残留量的测定高效液相色谱法》[6]用乙酸乙酯提取,正己烷液液萃取,MCX 固相萃取小柱净化,HPLC 紫外检测,外标法定量。以上方法中,DB33/T 693—2008《动物源性食品中硝基咪唑类药物残留量的测定高效液相色谱法》[6]为液相色谱法,只能检测甲硝唑等5 种硝基咪唑,检测限仅为2 μg/kg,灵敏度不高;GB/T 21318—2007《动物源性食品中硝基咪唑残留量检测方法》[4]和方力等[7]、黎翠玉等[10]的方法提取步骤较繁琐,实际操作中出现了提取液易乳化、旋转减压蒸馏蒸不干、过柱困难、标准曲线线性差、样品回收率低等问题;刘艳萍[8]、王志杰等[9]在乙酸乙酯中加入无水硫酸钠作为提取助剂,但无水硫酸钠容易吸附待测物质,影响提取效率,并且在提取剂浓缩后有盐结晶析出,对后续的质谱检测不利。
试验中发现二氯甲烷和乙酸乙酯对硝基咪唑类药物的提取效果较好,但二氯甲烷对人体毒性较大,不宜采用。由于硝基咪唑类药物是两性化合物,在弱酸性条件下以质子状态存在,在弱碱性条件下,以游离分子状态存在[11],因此考虑在提取剂乙酸乙酯中加入氨水。但加入的氨水太多会降低氯甲硝咪唑、洛硝哒唑的线性范围[12]。经试验发现,每15 mL乙酸乙酯中加入100 μL 氨水能够将洛硝哒唑、氯甲硝咪唑的线性范围提高至150 μg/L。由于提取剂乙酸乙酯与水互溶性较差,因此采用离心的方式分离上清中的水与乙酸乙酯。
硝基咪唑类药物的净化一般有液液萃取法和固相萃取法(SPE)2 种方式。液液萃取法净化操作相对简单,费用也较低,但净化效果不如固相萃取法。固相萃取法操作步骤繁琐,回收率受操作水平和固相萃取小柱质量影响较大且串联质谱检测的强抗干扰能力使其对样品的前处理净化依赖性很低。综上,本研究选择加入正己烷进行液液萃取净化。
2.2 仪器方法的优化
在水相中加入一定比例的甲酸能促进离子化[12],考察了不同含量的甲酸水溶液(0.10%、0.15%、0.20%、0.30%)对10 种目标化合物的分离效果,结果表明,0.10%、0.15%甲酸水溶液的分离效果均不如0.20%甲酸水溶液的分离效果,0.30%甲酸水溶液与0.20%甲酸水溶液的分离效果差别不大,为保护仪器,最后选择0.20%甲酸水溶液。流动相中加入甲醇会导致10 种硝基咪唑类药物出现不同程度的峰型变宽,灵敏度下降。选择0.20%甲酸水溶液-乙腈作为流动相,各目标化合物峰形尖锐对称,效果最好。
2.3 结果分析
按“1.3”项方法处理加标后的样品,10 种硝基咪唑类药物加标样品的离子流见图1,加标浓度均为10.0 μg/kg;阴性样品的离子流见图2,线性方程、检出限(LOD)和定量限(LOQ)见表5;分别按5.0、7.5、10.0、15.0、20.0 μg/kg 5 个加标浓度添加,每个水平测定4 次,回收率与相对标准偏差(RSD)见表6。结果表明,方法检出限为0.04~0.30 μg/kg,定量限为0.50~1.00 μg/kg,标准曲线线性范围为1~150 μg/L,相关系数(R2)≥0.998 00,回收率为73.0%~116.2%,相对标准偏差为0.43%~9.53%。
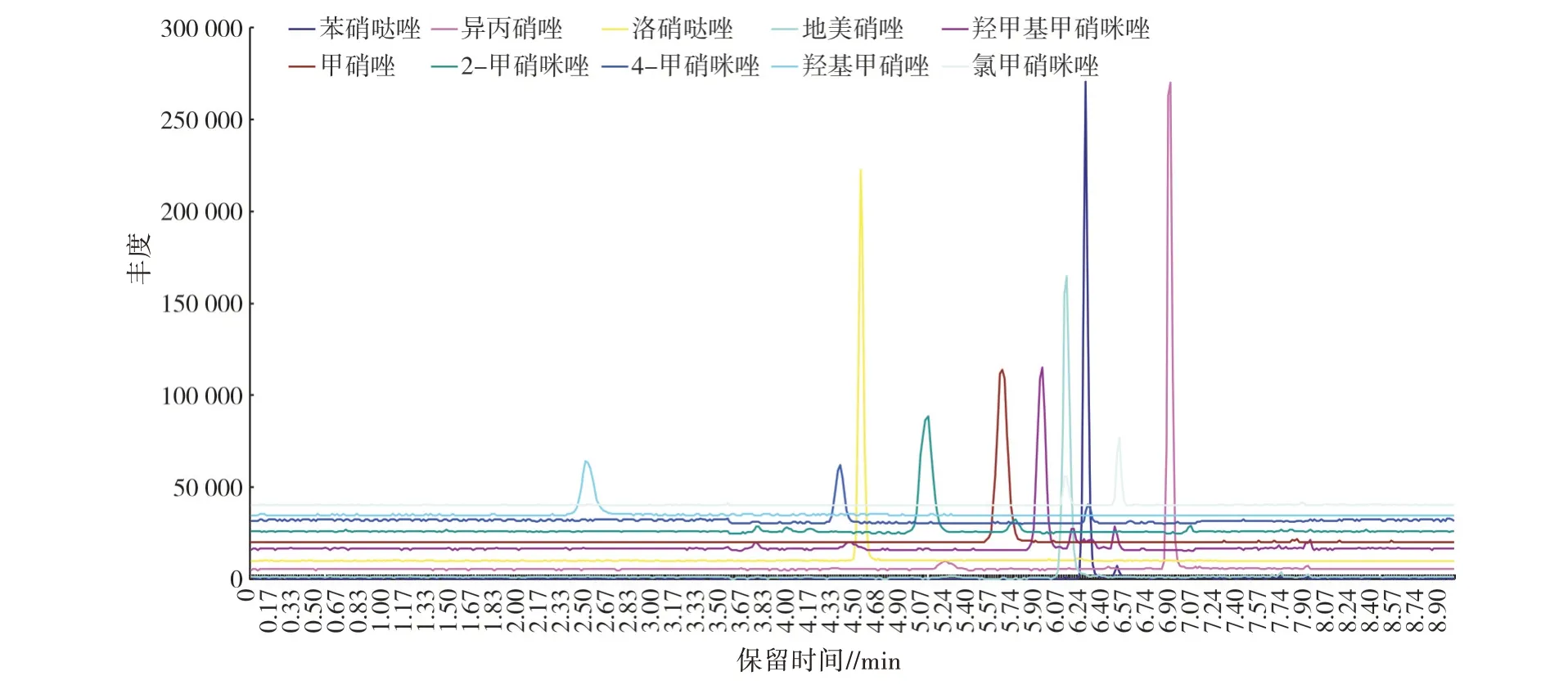
图1 加标样品的离子流
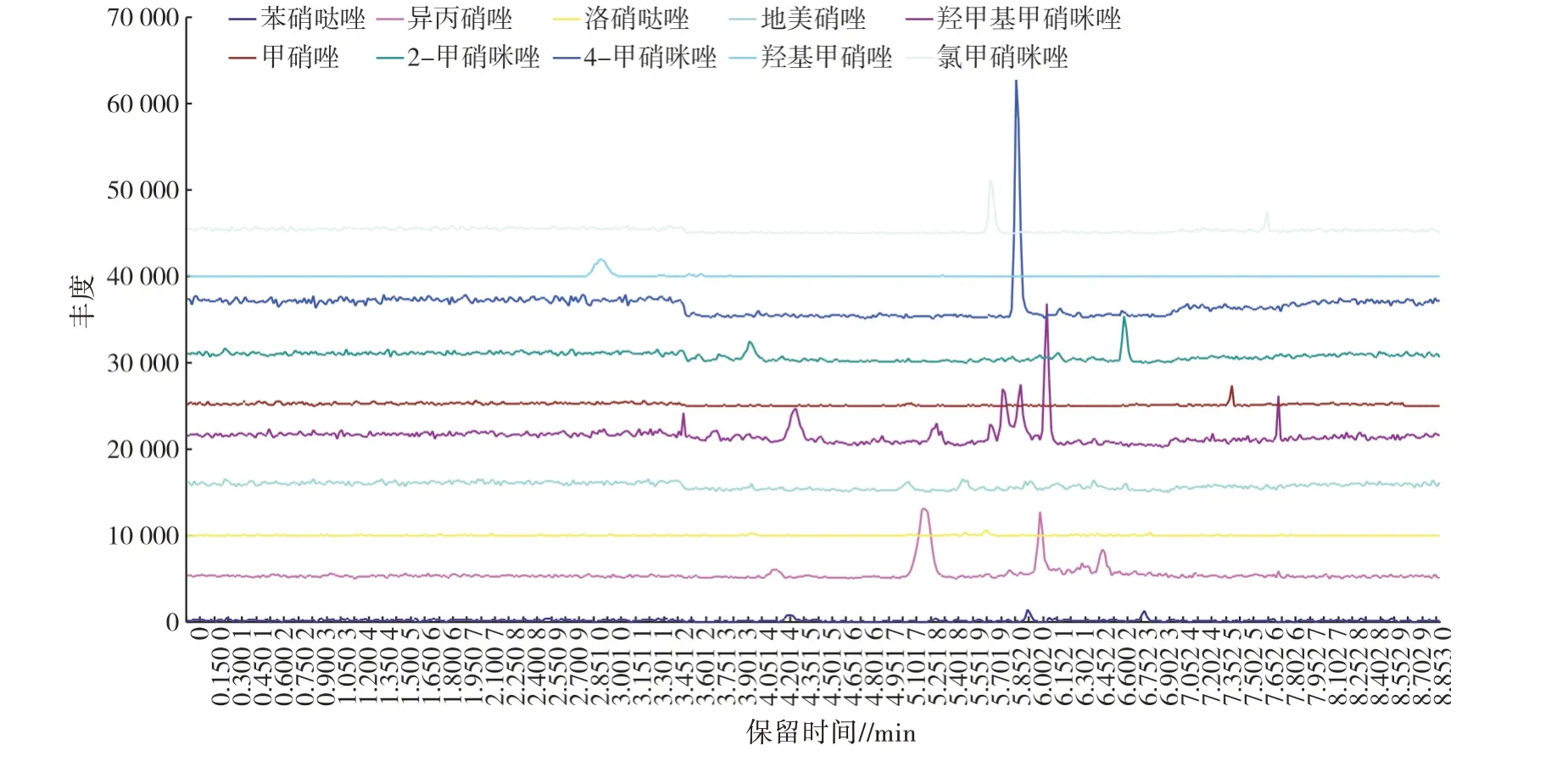
图2 阴性样品离子流
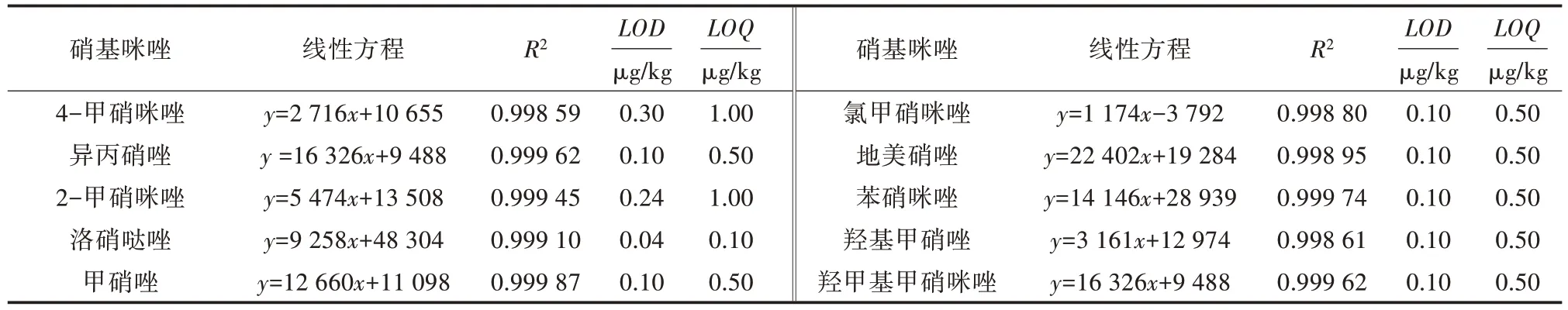
表5 10 种硝基咪唑药物的线性方程、检出限与定量下限
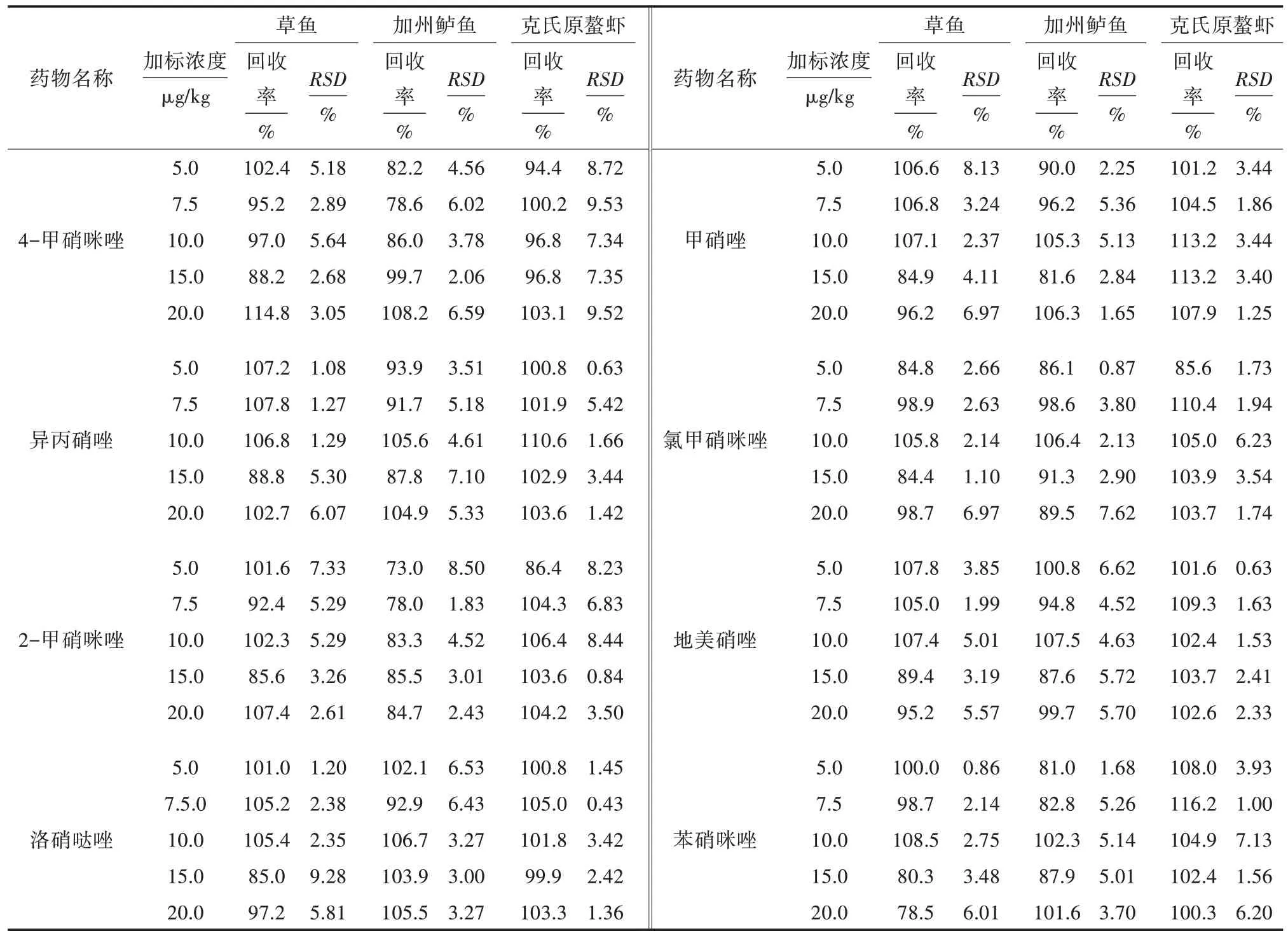
表6 方法的回收率和相对标准偏差
2.4 实际样品检测
利用本研究建立的方法对湖北省市售的4 批次共210 个水产品中10 种硝基咪唑类药物进行检测,结果发现,10 种硝基咪唑类药物在4 批次水产品中均未检出。
3 小结
本研究建立了同时测定水产品中10 种硝基咪唑类药物残留的高效液相色谱串联质谱法。标准曲线线性范围为1~150 μg/L,相关系数(R2)>0.998 00,在草鱼、加州鲈鱼、克氏原螯虾基质中分别添加5.0、7.5、10.0、15.0、20.0 μg/kg 硝基眯唑药物,回收率为73.0%~116.2%,相对标准偏差为0.43%~9.53%。该方法步骤简单、灵敏度高、重现性好,适合于大批量样品的检测。
