酶替代治疗法布里病的疗效分析
2023-11-02唐子洋李东昀邹玉荣李贵森陈莎莎
唐子洋 李东昀 张 萍 邹玉荣 李贵森 陈莎莎
[作者单位]1电子科学大学医学院(成都,610072);2四川省医学科学院·四川省人民医院肾脏内科
法布里病(Fabry disease,FD)是一种罕见的 X染色体连锁的隐性单基因遗传病,因GLA基因突变导致α半乳糖苷酶 A(α-Gal A)活性部分或全部丧失,造成其代谢底物三己糖酰基鞘脂醇(GL-3)及其衍生物脱乙酰基GL-3(Lyso-GL-3)在各组织器官中贮积,引起一系列脏器病变[1]。FD在正常人群中预估患病率为1/100 000[2-3]。国外报道新生儿中发病率约1/1 250~1/8 882[4]。其临床表现多样,常为神经、肾脏、心脏、皮肤、胃肠道、眼等多脏器受累,其中,肾脏、心脏、脑是后期主要受累脏器[5]。FD诊断需结合临床表现、酶活性、基因检测、生物标志物等多项指标,确诊需依靠酶学检查和基因检测[6]。患者多在青少年时期出现症状,随病程进展而加重,可出现心力衰竭、脑卒中及终末期肾病(ESKD)等严重并发症。男性患者预期寿命减少15~20年,女性患者减少6~10年[7]。酶替代治疗(ERT)使用重组α-Gal A替代体内缺陷酶治疗FD,在国外已有十余年的用药历史,2020-08-28获得中国国家药品监督管理局批准,用于确诊为FD患者的长期ERT,填补了国内治疗FD的空白[3,8]。目前国内尚缺乏有关西南地区ERT对FD疗效的报道。本研究通过分析四川省人民医院肾内科近1年治疗的8例FD患者的临床病理表现特点,首次报道了西南地区ERT的疗效及安全性,为FD的治疗提供医学经验和循证依据。
对象和方法
研究对象回顾性分析2022年1月至2023年6月四川省人民医院肾内科收治的8例FD患者。所有患者均签署知情同意书。
诊断标准和家系筛查依据《中国FD诊疗专家共识(2021 年版)》,结合患者临床症状、体征、家族史、α-Gal A 活性、基因变异证据和生物学标志物等明确诊断[2]。诊断标准:(1)具备GLA基因突变证据,α-Gal A[正常2.40~17.65 μmol/(L·min)]活性下降(女性患者可能正常),血浆 Lyso-GL-3(正常<1.11 μg/L)水平升高(女性患者可能正常);(2)具有FD相关临床表现如神经性疼痛、出汗障碍、皮肤血管角化瘤、角膜涡轮状浑浊、听力下降、肾脏受累和心肌肥厚等。7例患者接受家系筛查。
资料收集收集患者基本信息,临床表现,肾功能、尿液检查、血浆α-Gal A活性测定等,听力和眼科检查、心电图、心脏B 超、肾组织病理,基因检测,治疗反应等。治疗前和治疗半年后复查血浆 Lyso-GL-3、Mainz严重程度评分指数(MSSI)[9]、疼痛评分(BPI)。
基因检测采用 long range PCR方法,将GLA基因所有区域目的片段扩展,并纯化和建库,进行二代高通量测序。后经数据处理和分析,找出变异位点。
ERT方案[10]阿加糖酶α按照0.2 mg/kg静脉给药,每2周1次,根据临床症状轻重适当增减剂量。用药前30 min均给予异丙嗪抗过敏。用药过程中用心电监护仪监测生命体征,常备退热药、抗组胺药及氧气罩等抢救物品。
对症治疗针对患者脏器受累情况进行对症治疗。例4患者曾予血管紧张素转化酶抑制剂(ACEI)/血管紧张素受体拮抗剂(ARB)治疗尿蛋白3个月以上,例2曾使用激素半年后停药,例3、例6间断给予卡马西平、普瑞巴林治疗持续性周围神经痛。
统计学处理使用《SPSS 21.0》软件进行数据统计学处理。计数资料用例数或百分比表示;呈正态分布的计量资料以均数±标准差形式表示,采用独立样本t检验比较两组间的差异;不符合正态分布的计量资料以 中位数(四分位间距)表示,采用 Wilcoxon 秩和检验比较两组间的差异,配对检验比较治疗前后的差异。P<0.05为差异有统计学意义。
结 果
临床资料病例1:22岁男性患者,18岁时因胃肠不适,肢端疼痛,皮肤角质瘤,检查发现尿蛋白2+,尿蛋白定量4.505 g/d,白蛋白40 g/L,行肾活检电镜提示FD。病例2:26岁女性患者,19岁出现肢端疼痛,双手掌侧、颜面部可见皮肤血管角化瘤,蛋白尿2+,外院给予激素等免疫抑制剂,疼痛和尿检无缓解,22岁行肾活检和基因检测确诊。病例3:21岁男性患者,11岁出现皮肤血管角质瘤,四肢疼痛,无肾脏、心脏、头颅等受累,多家医院皮肤科、神经科就诊,给予卡马西平、普瑞巴林等无缓解,1年前基因检查确诊。病例4:47岁男性患者,42岁时体检发现血清肌酐(SCr)升高94 μmol/L,蛋白尿3+,血压正常,肢端疼痛,皮肤血管胶质瘤,给予ACEI/ARB治疗无缓解,45岁行肾活检电镜胞质内见大量环状髓样小体。病例5:65岁女性患者,7年前因胸闷、胸痛、蛋白尿、肢端疼痛住院检查心脏彩超提示左房,右房增大。主动脉瓣钙化。头颅MRI提示多发性腔隙性脑梗死,脑萎缩。SCr 112 μmol/L,尿蛋白定量1 957 mg/d,行基因检查确诊。病例6:26岁男性患者,6岁出现间歇性肢端剧烈疼痛、感觉异常,排汗减少,无肾脏、心脏、头颅等受累,给予卡马西平、普瑞巴林等无缓解,20岁行基因检查确诊。病例7:47岁男性患者,7岁出现双足趾疼痛,颜面部及双下肢水肿伴心累气紧,42岁出现双上肢疼痛,水肿、心累加重、高血压,当地医院诊断“慢性肾衰竭、尿毒症、心力衰竭、房室传导阻滞”,行起搏器置入术,规律透析,透析后疼痛可缓解,46岁出现视力、听力、记忆力下降,心超提示左室收缩舒张功能重度减低基因检查确诊。病例8:57岁男性患者,10岁出现肢端疼痛,无汗,皮肤血管角质瘤,20岁发现蛋白尿、SCr升高600 μmol/L,水肿、心累、气紧、少尿、血压升高,彩超提示肥厚性心肌病,听力减退,腹痛/腹胀/腹泻,30岁行肾移植,40岁SCr升高700 μmol/L,规律透析,57岁基因检查确诊(表1)。

表1 8例Fabry病患者基线临床表现特征、酶替代治疗用药次数及不良反应

表2 8例法布里病患者基线影像学检查
实验室及影像学检查8例患者均出现α-Gal A活性降低,lyso-GL-3升高。4例患者有蛋白尿,4例患者肾功能受损(2例ESKD,其中1例行肾移植20年后再次透析),6例患者心脏彩超级心电图异常,3例患者心功能受损,1例脑梗死,脑萎缩,2例听力受损,2例胃肠道功能紊乱,1例可疑肿瘤,8例均未见角膜涡状浑浊,无肺通气功能异常(表1、2)。
基因检测及家系筛查8例患者基因检测显示,7个GLA基因错义突变,1个无义突变。1个剪切突变,1例无家系调查(例6),6例基因突变来源于母亲,例8来源不明(图1)。
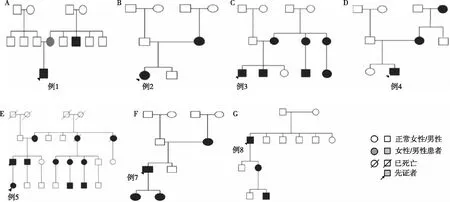
图1 7例法布里病患者家系图
肾脏病理3例患者(例1、例2、例4)行肾活检。甲苯胺蓝染色显示足细胞质内含大量嗜甲苯胺蓝的蓝色颗粒状物;电镜下见肾小球足细胞肿胀,足细胞弥漫空泡变性、蜂巢样改变,胞质内可见大量嗜锇性、同心圆样的髓样小体或斑马小体,多数患者小管间质中重度病变。肾间质血管病变突出(图2)。

图2 A、B:肾小球足细胞空泡改变(A:HE,×200;B:PAS,×400);C:足细胞内嗜甲苯胺蓝的颗粒状物质(甲苯胺蓝染色,×400);D~F:足细胞胞质内充满嗜锇性“髓样小体”(EM)
治疗反应8例患者中位随访时间10.5月(7.2~12.7月),接受阿加糖酶α治疗4~22次,8例肢端疼痛的患者治疗后BPI评分及MSSI量表评分明显降低(P<0.001)。4例患者蛋白尿较前有明显下降(P=0.037),其中有3例既往未接受ACEI/ABR治疗,在接受ERT同时开始给予ACEI/ARB治疗3月以上,另1例曾接受3月以上ACEI/ABR治疗,叠加使用达格列净,4例蛋白尿患者均改善生活方式同时接受ERT,血压均控制在120/80 mmHg以下。7例患者治疗半年后复查血浆 Lyso-GL-3水平较基线值显著下降(P=0.01)。输注阿加糖酶α过程中均未发生皮疹、血管神经性水肿、血压降低等不良反应及其他严重不良反应(表3)。
经典型和迟发型的对比经典型组患者的发病年龄、α-Gal A活性较迟发型低,Lyso-GL-3水平较迟发型高,符合FD表现特点(表4)。

表4 经典型组和迟发型组法布里病患者的临床表现及实验室检查结果
讨 论
FD可导致肾衰竭、心肌肥厚、四肢神经痛、角膜涡轮状浑浊等,严重者出现心脑血管并发症或ESKD,甚至过早死亡。目前关于中国 ERT 的报道尚不多,仅有两项关于阿加糖酶α在FD患者中的治疗情况[11-12],且皆来自中国东部,本文首次报道了西南地区ERT的疗效及安全性,为FD的治疗提供循证依据。
国外文献报道迟发型发病率比经典型高出10倍[13],据瑞金医院统计显示,国内目前诊断的FD的临床分型 66.1%男性患者为经典型,75%女性患者为迟发型[14]。本研究中经典型FD患者男性占比较多,经典型组患者的发病年龄、α-Gal A活性较迟发型低,Lyso-GL-3水平较迟发型高,男性α-Gal A活性严重下降或缺失,大多数女性患者在正常范围,男性患者病情更重,女性患者症状相对较轻,符合FD表现特点和国内外既往研究[12-13]。本研究7例患者经过6月ERT后复测血浆Lyso-GL-3水平均显著下降,与临床表现疼痛减轻、生活质量改善等疗效结果一致[1]。FD患者的疼痛主要为神经性疼痛,多表现在四肢肢端,且患者肢端疼痛早于肾脏病之前,童年开始就出现脚趾疼痛,伴无汗[15]。本文8例患者均有反复发生肢体疼痛,程度严重影响生活,一般止痛药物效果差,多个科室就诊,治疗效果不佳,追问病史及家族史,α-Gal A明显下降,最终确诊FD。提醒肾脏疾病合并肢体疼痛无汗、少汗,心律失常、心肌肥厚,有家族史时需警惕FD,筛查家族中有无相似症状者。
肾脏受累的FD患者通常预后较差,约30%的患者在30岁左右进展至ESKD[15]。本研究6例患者出现肾脏受累,表现为蛋白尿、血尿、SCr升高,3例患者行肾活检示足细胞内见大量髓样小体。4例(例1、例2、例4、例5)患者蛋白尿较前明显下降(P=0.037),但不能除外生活方式、ACEI/ABR、钠-葡萄糖协同转运蛋白2抑制剂(SGLT2i)的影响,3例肾功能受损患者(例5、例7、例8)发现心力衰竭,心电图、心脏彩超异常,经ERT及透析治疗目前心功能较前改善。2 例男性患者分别在42 岁、20岁因进展至ESKD期而开始透析,1例在30岁行肾移植手术,术后10年再次进入透析。与既往研究结果一致,肾脏替代治疗或肾移植均不能有效提高其生存率[16-17]。强调早期诊断,早期干预。心脑血管系统受累对预后有重要影响,但检出率低,临床需加强筛查。虽然基因检测为FD确诊的金标准,但FD肾脏表现不具有特异性,肾活检和酶活性测定对该病的诊断则起到关键作用[18],且较基因检测更为简便。因此,肾活检(尤其是电镜超微结构检查)对FD的诊断尤为重要[15],肾内科医师首先应对该病有深入的了解和认识。
FD发病年龄、进展速度和器官损伤的表现、突变基因在不同家系中存在较大的差异。国外一项研究报道在75例先证者家系筛查中共确诊FD373例,分析发现,平均每个先证者有5个家庭成员被诊断为FD[19]。本文中每个先证者有2~11个家庭成员被确诊,同一家系中男女表现不同,如例8先证者皮肤、肾脏、心脏、听力均受损,其孙子在5月龄即有皮肤、周围神经受累,但其女40岁经基因确诊为FD,目前无特殊不适。例1先证者累及皮肤、肾脏、神经三大系统,检出一错义突变LMXIBc.722C>G(p.Ser241Trp)无文献报道,但文献报道在1例甲-髌综合征患者中检测到有影响同一个氨基酸残基的错义突变c.721T>C(p.Ser241Pro)。其余6例患者的错义突变均为既往报道的致病突变,可能对蛋白正常功能有害。目前尚未发现GLA基因突变与临床表型之间有显著相关性。
FD的治疗包括非特异和特异性治疗[14,18-20],即对症治疗和ERT。对症治疗主要对各脏器受累情况给予相应的对症治疗(如止痛、ACEI/ARB、透析、移植、心脏起搏器等)。特异性治疗包括ERT、分子伴侣疗法/酶增强治疗、底物减少治疗、基因治疗及基于mRNA的治疗[21]。ERT作为外源性补充基因重组的α-Gal A,替代患者体内缺失的α-Gal A,从而促进 GL-3的分解,减少GL-3和Lyso-GL-3在器官组织的贮积。ERT开始越早患者受益越显著[4,22-23]。FOS研究是一项国际大型FD结局调查研究[19],于2001年启动,主要关注FD患者的临床结局,瑞普佳治疗的长期安全性和疗效。该研究纳入既往接受阿加糖酶α治疗≤10年的FD患者560例,评估基线时心脏肾脏不同受损程度对ERT疗效的影响,结果显示在整个随访期间,基线时左心室质量指数(LVMI)异常的患者心血管事件风险显著更高。在整个随访期间,基线时估算的肾小球滤过率(eGFR)异常的患者肾脏事件风险更高[24-25]。目前我们已治疗8例FD患者,所有患者治疗期间未见严重不良反应,治疗后肢端疼痛症状、生活质量明显好转,蛋白尿、肾功能都保持相对稳定,心功能稳定,BPI、MSSI评分明显下降。我们的短期疗效研究证实了阿加糖酶α治疗可显著缓解神经性疼痛,心肾功能的改善有待远期进一步研究,在后续随访过程中完善eGFR 斜率、LVMI对比及MSSI评分对比,加强多学科合作,及高危人群筛查,提高FD疾病的诊治。综上所述,FD的诊断需结合临床表现、酶活性、基因检测、生物标志物等多项指标,确诊需依靠酶学检查和基因检测,ERT为FD患者的治疗基石。短期阿加糖酶a治疗未见严重不良反应,治疗后患者血浆Lyso-GL-3水平下降,临床症状明显改善,但疗效具有剂量依赖性,临床获益需要更长时间的观察和随访。
