机器学习在化学合成及表征中的应用
2023-10-30李子昊张书宇
孙 婕, 李子昊, 张书宇
(上海交通大学 化学化工学院,上海 200240)
近年来,科研工作者一直在寻找能够快速、简捷、高效发现药物、催化剂、蛋白质等新材料和新反应途径的自动化合成系统.人工智能(Artificial Intelligence, AI)和机器学习(Machine Learning, ML)的出现使实现这一目标成为可能.ML是AI和计算机科学的一个重要分支,它研究和构建的是一种特殊算法而非某一个特定的算法,能够让计算机自身从数据中学习并进行下一步预测[1].ML算法能够从大量化学数据中寻找规律和联系,帮助科研工作者做出更合理的判断和决策, 加快研究过程.ML在化学合成领域[2-3]的应用已经取得许多令人瞩目的成果,如分析化学反应进行反应优化[4-6]、逆合成分析寻找产物的最佳合成路径[7-8]、比较药物活性辅助药物设计[9]等.ML正在成为除分子模拟之外的计算化学的新范式.ML为化学合成领域的发展带来无限生机的同时,也为合成研究带来了新的难题与挑战.
1 ML应用于化学合成的基本流程
ML为计算机系统提供自动学习和增强经验的能力,并且无需专门编程,被称为第四次工业革命中最流行的技术[10-11].ML通过训练算法查找数据之间的相关性,并根据该分析做出最佳决策和预测.基本思路是将实际问题抽象成数学模型,利用数学方法对模型进行求解,最后采用指标对模型进行评估.因此,可以将ML在化学合成领域的应用简化为如图1所示的4个步骤:建立反应数据集、特征化数据、训练模型和分析结果.

图1 在化学科学中应用ML算法的工作流程
1.1 建立反应数据集
ML模型需要海量数据作为支撑,借助数据进行模型训练.一般而言,可以借助当前已公开的化学数据库如SciFinder、Reaxys、USPTO等[12]进行初步筛选,但科研工作者无法直接获得批量数据.可用数据集往往需要耗费一定时间进行筛选、汇总和整理,科研工作者需将其按照7∶3或8∶2的比例进行划分,训练集数据占多数.需要注意的是不能使用所有数据进行模型训练,也不能使用训练集数据对模型进行评估.但允许所有数据进行特征化之后再进行数据集划分.
1.2 特征化数据
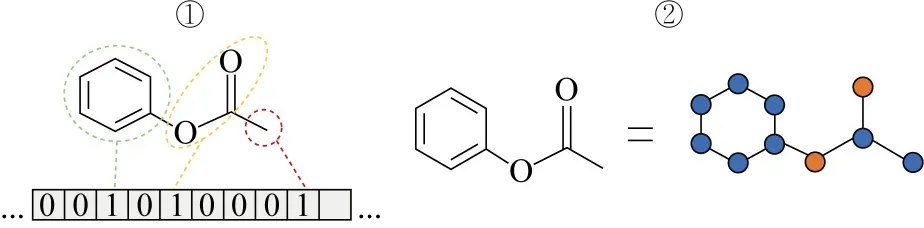
输入数据的形式往往影响ML的效果,常见的输入数据形式主要有向量、矩阵和图像3种.由于化学数据的特殊性,绝大多数数据无法直接作为模型的输入,需要进行数据转换.将原始数据转换成更适合算法处理的格式,这一过程称为特征化或特征工程.在化学领域中,使用分子描述符对分子信息进行描述表示.分子描述符是指分子在某一方面性质的度量,既可以是分子的物理化学性质,也可以是根据不同算法推导出来的与分子结构相关的数值指标.选择出与研究对象最密切相关的描述符对整个ML过程而言十分重要,表1总结了常见的分子描述符.

表1 常见的分子描述符总结(以苯酚乙酯为例)
其中,需要特别注意量子化学描述符,这类描述符一般通过Gaussian、NWChem等软件计算获得,能够较准确地描述分子的化学和物理性质,计算结果具有较高的可靠性.此类描述符不仅是ML的输入数据形式之一,而且能够作为化学实验结果的佐证工具,如张书宇等[15]总结了密度泛函理论(Density Functional Theory, DFT)计算验证轴向手性苯乙烯合成的机理和方法.首先,他们发现镍催化对映选择性三分量自由基传递烯烃还原偶联可以实现轴向手性苯乙烯的合成,可以借助DFT计算寻找反应中的过渡态对整个反应机理进行佐证[17].其次,使用DFT计算非常规远程杂芳基迁移对非活性烯烃进行异芳基氟烷基化过程中杂芳基迁移后氢原子转移(Hydrogen Atom Transfer, HAT)的溶剂化自由能,计算结果与实验数据吻合,与反应机理相印证[18].
1.3 模型训练
数据特征和算法性能决定了ML的有效性和正确率,不同的ML算法产生的结果不同,甚至同一种ML算法结果也会因数据特征而异.算法是ML过程的关键,选择时需从实际问题出发,多方面综合考虑.目前,根据算法特征将ML分为4类[19]:监督学习(Supervised Learning, SL)、无监督学习(Unsupervised Learning, UL)、半监督学习(Semi-Supervised Learning, SSL)和强化学习(Reinforcement Learning, RL).
SL是目前使用最广泛的ML方法,用于学习从输入映射到输出的函数f(x),f(x)为每个输入x产生的输出y或给定x的y上的概率分布[1, 20].当训练数据为离散型数据时采用分类算法,为连续型数据时采用回归算法.SL要求训练数据为带有“标签”的数据,常见算法包括支持向量机(Support Vector Machine, SVM)[21-23]、K-均值聚类(K-means Clustering)[24]、线性回归( Linear Regression, LR)[25]、逻辑回归( Logistic Regression, LR)[26]等.
UL训练未标记的数据集,无需人工干预,可以理解为数据驱动的过程[20].UL本质是一个统计手段,其输入数据没有被标记且结果未知.该算法的目的不是向计算机施加指令,而是让计算机自主学习,促进ML向自动、灵活和通用方向发展.如今使用较多的UL算法是聚类算法,聚类是将相似的对象分到不同的组中,或者更准确地说,将一个数据集划分为子集,从而使每个子集中的数据根据某种定义的距离度量[27].
SSL巧妙地将SL和UL结合在一起.在训练期间,它使用少量拥有标签的数据集来指导大量未标签化数据集进行分类和特征提取.SSL可以解决带标签数据不足或无法负担标记足够数据的费用而不能进行SL的问题.
RL不要求预先给定任何数据,而是通过接收环境对动作的奖励(反馈)获得学习信息并更新模型参数[28].RL的思路非常简单:如果在一件事中采取某种策略可以取得较高得分,那么就进一步“强化”这种策略,以期取得更好的结果.可以认为,RL是所有形式的ML中最接近人类和其他动物学习的方法,也是目前最符合AI发展终极目标的方法.RL系统一般包括策略、奖励、价值和环境/模型4个要素.2016年AlphaGo击败世界围棋大师李世石[29]和2018年谷歌训练机器臂的长期推理能力[30]等是RL应用的最佳佐证.
1.4 分析结果
待模型训练完,可以将真实实验数据与预测数据进行对比来评估模型质量,分析模型学习结果能否较好解决实际问题.针对不同问题,需采用不同的模型评估指标,如评估SL中分类模型可以采用准确率(Accuracy)、召回率(Recall)、受试者工作特征曲线 (Receiver Operating Characteristic, ROC)等;评估SL中回归模型时可以借助平均绝对误差(Mean Absolute Error, MAE)、均方误差(Mean Square Error, MSE)、均方根误差(Root Mean Square Error, RMSE)、决定系数(R2)等.
2 ML在化学合成及表征领域的应用
ML在化学合成领域最早的应用可以追溯至20世纪60年代,Corey等[31]开发了基于规则的计算机辅助合成设计程序(Computer-Aided Synthetic Planning, CASP),该程序辅助化学家快速实现化合物合成,输入分子结构信息,输出不同的反应合成方案.在过去的几十年间,随着计算机硬件设施的更新和大型化学数据库的建立,ML在化学合成领域的应用日益广泛.文献[32-33]中从化学家的角度介绍ML相关应用,本文从ML算法的角度,介绍化学合成及表征领域中如何使用ML模型.
2.1 随机森林的应用
随机森林(Random Forest, RF)是一种集成分类算法,由 Breiman[34]提出,使用“并行”决策树(Decision Tree, DT)的方式,如图2所示.DT模型是一种以树结构为依据的分类算法,由节点和分支组成.从树的根节点开始,依次向下分类.一棵DT有且仅有一个根节点.能够将一个复杂的决策过程分解成一组更简单的决策,从而提供一个通俗易懂、易解释的解决方案是DT模型最大的优势[35].在RF中,每棵DT生成一个随机向量,向量之间相互独立且分布相同,根据一定的投票机制或取平均值得到最佳分类结果.RF由多棵DT组合生成的,因此该算法能够最大限度地减少过拟合问题,提高预测精度和控制力[36].
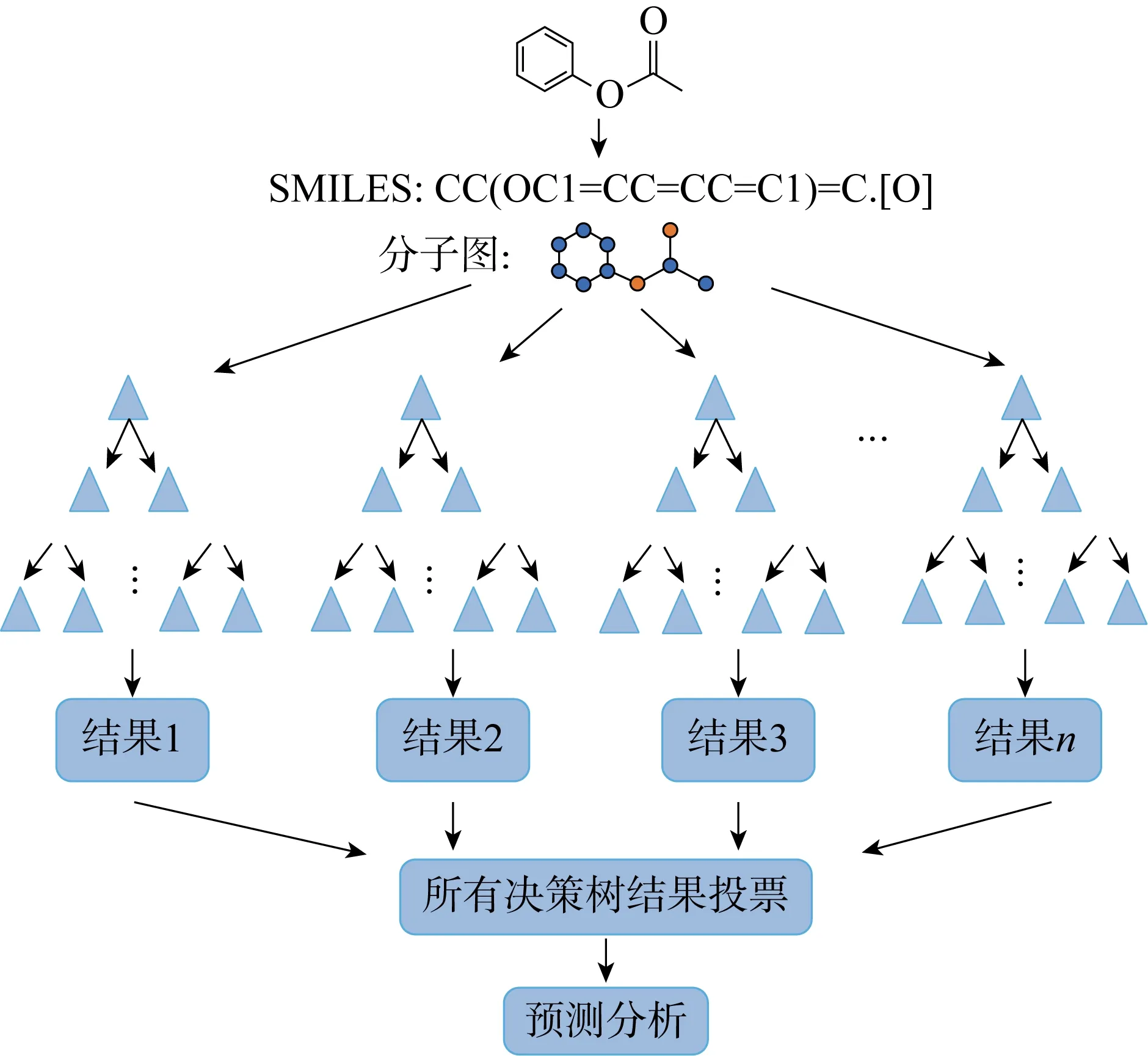
图2 输入数据形式以分子图和SMILES字符为例的RF模型
RF作为一种集成算法,具有良好的鲁棒性和可靠性,分类效果好,适合作为基线模型进行产率预测.高通量实验结合DFT计算能够缩短RF模型建立的时间,使得该模型在化学合成领域得到广泛应用.RF解决的主要是分类问题,分类问题是数据挖掘处理的一个重要组成部分,目标是根据已知样本的某些特征,判断新样本属于哪种已知的样本类.科研工作者通常从预测精确度、计算复杂度、模型简洁度对多种分类算法进行比较评价.Singh等[35]选择了5种不同的轴向手性联萘催化剂共368个不对称氢化反应和一系列烯烃、亚胺作为训练数据集生成了一个RF模型,如图3(a)所示,选择能够共享等效或具有共同核心区域的反应参数作为输入,以对映体过量百分率(ee%)作为输出值,对输入化合物进行分类,每种催化剂生成一棵DT,形成RF模型.与其他模型相比,RF模型得到了较高的精度,表明RF模型在识别不对称催化反应时有良好的应用.Kang等[37]设计了一种RF模型用于预测分子的激发能量和相关振荡器强度,首先使用RDkit工具包计算出分子的扩展连通性指纹(Extended-Connectivity Fingerprints,ECFP)、MACC键等分子描述符作为输入数据生成许多DT模型,对所有DT的预测结果进行投票选择评估,生成RF模型.该模型使用了近50万个DFT数据进行训练,实验结果表明RF模型预测振荡器强度和有机化合物最高强度跃迁激发能的精准度最佳.Li等[38]报道了一种物理有机特征描述符和RF相结合的模型(PhyOrg-RF),对杂环自由基C—H官能团的区域选择性进行预测.在样本外测试集中PhyOrg-RF模型实现了94.2%的位点预测精度和89.9%的选择性预测精度,拥有较好的区域选择性预测能力,使用其他已公开实验数据进行测试验证了PhyOrg-RF具有优异的泛化能力.Ahneman等[39]提出一种基于RF的预测钯催化Buchwald-Hartwig胺化反应产率的模型,如图3(b)所示.其中,10 mol%表示催化剂与反应物的物质的量之比为10%.通过高通量实验生成 4 608 个反应数据,将简单原子、分子和振动描述符作为训练集进行模型训练.该模型的测试集RMSE为7.8%,R2为0.92,该模型未曾出现过拟合现象,能够以RMSE为11.3%、R2=0.83的精度成功预测反应产率.Tomberg等[16]选择RF作为分类模型,判断芳香类化合物的反应位点,与人工神经网络(Artificial Neural Network, ANN) 、LR和SVM模型相比,RF不仅训练时间短,而且正确率高达93%.Xu等[40]提出一种将过渡态知识模型与额外树(Extra Tree, ET)模型相结合的方式,对钯电催化C-H活化的对映选择性预测.RF在一个随机子集内得到最佳分类属性,而ET完全随机得到分类属性,同时具有随机性和最优性.

图3 催化剂分类形成RF模型过程和RF作为比较模型的反应产率预测
2.2 卷积神经网络的应用
神经网络(Neural Network, NN)指包含多个阈值单元的多层网络,每个阈值单元实现不同的简单功能,将每个单元的结果进行汇总得到输出结果,根据优化算法调节整个网络的参数实现网络最优.卷积神经网络(Convolutional Neural Network, CNN)是将卷积核和NN相结合的一种算法.自LeCun等[41]提出LeNet-5多层ANN,CNN逐渐出现在人们视野.CNN由卷积层、池化层和全连接层组成,如图4(a)所示.卷积层由卷积核组成,用于生成特征图.根据下式可以求出第k层第n个特征图中(i,j)处的特征向量:
(1)
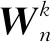
CNN主要用于解决SL中的回归问题.回归问题研究的是自变量和多个变量之间的关系,用于处理离散型数据.NN受人类大脑的启发,模仿生物神经元信号相互传递的方式,能够无限逼近非线性模型,在化学合成领域有着出色的表现.首先,CNN借助共享卷积核的方式降低计算复杂度,可以快速处理高维化学数据.Hirohara等[47]设计一种SMILES字符串与CNN相结合的模型(SCFP),用于化学基序检测.使用TOX21数据集中分子的SMILES字符串,将字符串输入CNN中,其中卷积操作只在SMILES字符串的一个方向进行,如图4(b)所示(k1和k2表示滤波器),由受试者工作特征及其曲线面积(ROC-AUC)进行评分.此模型还可以被视作一种分子指纹,在SR-MMP子数据集的化学空间中比ECFP分子指纹表达效果好.选取NR-AR子数据集进行化学基序分析成功检测出一种类固醇样化学基序.Wallach等[48]报道了一种基于CNN的模型AtomNet,预测药物发现应用中小分子的生物活性.AtomNet有两点优势:①CNN的强制局部性与化学基团之间相互作用时产生的局部效应相吻合;②将有关配体的信息和相关目标结构的信息相结合,十分适合结构的亲和力预测,并且选择原子在靶位结合点的位置,能够让模型发现任意分子特征.选择文档理解数据集和评估(DUDE)基准的数据集,评估数据集的AUC及其对数值,AtomNet中 57.8%目标的AUC大于0.9.Hughes等[49]使用702个环氧化反应数据训练了一种CNN模型,在环氧化位点识别上表现出0.949 的AUC结果,在区分环氧化分子上表现出0.793 的AUC结果.此网络不仅能够预测分子的环氧化作用,还能预测分子中的环氧化作用位点.该课题组还将类似的模型应用到了小分子与软亲核试剂的反应预测中,对是否能够发生反应进行预测,准确率为80.6%,小分子反应位点的预测准确率达到了90.8%[50].
CNN不仅可以对高维数据进行快速降维,在图像处理如图谱分析方面也有不可比拟的优势.Xing等[51]提出一种基于CNN的生物学驱动代谢组学习工作流程SteroidXtract,可实现在非靶向代谢组学数据集中对类固醇化合物二级质谱谱图(MS2)的自动化快速索取.SteroidXtract是一种高灵敏度、高特异性提取类固醇化合物谱图的工具,该方法不使用传统统计驱动的代谢组学习数据处理过程,更加高效简洁.Zheng等[52]借助CNN得到一种快速获取高质量核磁共振纯位移谱的新方法,如图5所示.通过在实验中引入指数采样来加速PSYCHE纯位移谱的获取,使用CNN对欠采样的图谱进行重建,可以在低采样率的情况下获得干净的纯位移谱.卷积核的选择对CNN算法的成败有着关键作用,通常选择大小为3×3,步长为1的卷积核.此外,也可根据实际应用进行调整,但需注意的是,卷积核尺寸越大、步长越大,得到的特征图数量越少,提取出的特征数目越少,可能会影响后续预测的准确性.共享卷积核使得CNN算法复杂度大大降低,因此当处理大量高维数据时,推荐使用CNN算法.

图5 CNN采集核磁共振纯移位波谱[52]
2.3 图神经网络的应用
几何深度学习[53]的出现将NN模型扩展到了非欧氏空间.图神经网络(Graph Neural Network, GNN)是处理非欧氏空间数据的常用模型,能够以递归形式合并邻近节点的信息或消息,同时自然地捕获图形结构和节点特征[54].GNN通过图节点之间的消息传递捕获图中重要信息, 查看相邻节点上的信息来确定每个节点的最终状态,以迭代方式传播相邻节点信息来学习目标节点的特征,直至到达稳定的固定点.简单来讲,GNN获取信息的过程可以概括为:聚合—更新—循环,如图6所示.首先使用某种方法对节点信息f1~f5进行表征描述,使每一个节点学习一个嵌入状态,这个状态用来产生所需要的输出即更新后的节点信息f1,new~f5,new.给定节点和边的特征即可不断更新节点状态并获得最终输出.当所有节点的状态都趋于稳定状态时,节点的状态向量都包含了其邻居节点和相连边的信息,需要保证整个更新过程收敛.

图6 GNN信息更新过程
GNN通过信息传播机制不断交换邻域信息以达到更新节点状态的目的.但GNN不能无限次更新节点的状态向量,会出现过拟合现象.为解决该问题,2016年Li等[55]提出门控图神经网络(Gated Graph Neural Network, GGNN)算法.GGNN引入门控递归单元进行循环迭代,能在一定程度上防止过拟合现象.随着NN的发展,Kipf等[56]提出图卷积神经网络(Graph Convolutional Neural Network, GCNN),GCNN使用NN作为更新函数,每层可以使用不同的更新函数,有效提高更新程度.
相比众多算法的输入数据形式,GNN的分子图形式能够在一定程度上减少化学数据特征的丢失.Duvenaud等[57]开发了一种基于GNN的神经图指纹,采取可微操作代替圆形指纹的离散操作,单层NN取代哈希结构,具有以下优势:①在溶解度、药物功效、有机光伏效率等性能中比固定指纹好;②仅编码能区分相关特征的部分,使用数据量少;③每个特征都可以被相似但不相同的分子片段激活,可解释性强.Coley等[58]将预测反应物的性质看作基于图的任务,输入反应物分子的分子图进行训练,生成Weisfeiler-Lehman神经网络(Weisfeiler-Lehman Netwrok, WLN),分析反应物图并预测原子对更改每个新键序的可能性,准确率超过85.6%,每个反应耗时约100 ms.同时该课题组还选择11位人类化学家与该模型进行对比,发现该模型非常适合用于寻找新分子.Saebi等[59]将化学反应表示为分子图,用GNN和反应化合物的化学性质作为框架预测反应性能.输入结构和空间特征获得产量分数,取分数的平均值来生成反应产量预测.验证Suzuki-Miyaura数据集得到的R2为0.962±0.010.
分子图表示形式有两个关键优势是旋转不变性和平移不变性,在化学合成领域中可以加速计算模型的建立.化学中的计算模型旨在使用基于量子化学的计算来确定给定分子系统的性质和行为[60].Roszak 等[61]开发了一个基于GCNN的酸解离常数(pKa)预测器,实现了在毫秒级时间内准确预测C—H酸的pKa,对13 000个反应的预测达到了90%以上的正确率.直接使用节点嵌入预测pKa所有原子的值进而寻找分子中酸性最强的质子.GCNN可以提供快速准确的原子特异性特征预测.该GCNN模型还在其他实例中展示出90%的预测反应位点正确率,显示出其在合成规划中的潜在应用.Wen等[62]设计了BondNet模型,用GNN预测键离解能,在自行构造的中性和带电荷分子的均溶、异溶键离解能数据集和PubChem的数据集上,MAE分别为0.022、0.020 eV,显著低于化学精度 0.043 eV.Grambow等[63]开发了一个基于GNN的模型来预测给定的反应物活化能,并在一个新的、不同的气相量子化学反应数据集上训练该模型,结果表明该模型实现了准确的预测并且符合对化学反应性的直观理解.
2.4 UL和RL的应用
UL常用于特征处理,将高维特征进行降维,如主成分分析法(Principal Component Analysis, PCA)[64]对初始特征线性组合生成新特征,将不相关的新特征按方差进行递减排序,减少特征数量从而加快ML模型建立.Zahrt等[64]采用PCA对数据进行降维,对化合物的高维空间进行降维,保留使数据方差最大化的新维度,选择代表性子集进行相关预测分析.
RL在逆合成设计中采取不确定性下的决策,不仅比传统方法处理速度快,而且置信度更高.RL通常以马尔可夫决策过程为框架[65],奖励函数为核心,奖励函数决定了主体通过动作学习实现的目标.奖励函数可以是离散的也可以是连续的.Segler等[66]设计了一种将蒙特卡洛树和NN相结合的逆合成分析方法,由计算机辅助合成设计程序(Computer-Aided Synthetic Planning, CASP)生成合成路线,使用蒙特卡洛树和3个不同的NN进行搜索.从目标分子开始,选择树中最有可能的下一个位置,直至到达叶节点.通过扩展策略预测可能出现的叶节点的子节点,并将其添加在树中,对推出过程进行评估.结果的位置值表示RL更新其树搜索策略所需要的奖励,找到解决方案会收到奖励,找到部分解决方案会收到部分奖励,未找到方案则会收到惩罚.不断迭代更新直到达到最大的时间或迭代次数,通过选择具有最高位置值的断开路径来决定最终的合成路线,具体搜索过程如图7所示,包括选择最可能的位置、使用扩展程序对节点进行扩展、选择评估新节点和更新4个阶段,其中T1~Tn为所有可能的概率分布,R1~Rk表示完整的反应物.
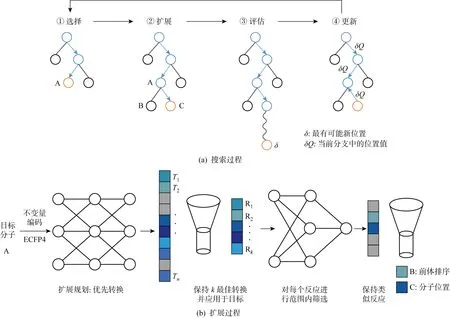
图7 蒙特卡洛树搜索的4个阶段[66]
3 挑战与机遇
3.1 建立高质量数据集和模型评估标准
数据集质量直接决定了ML模型训练的成败,构建大型数据集是一个耗时且费力的过程,因此这些数据集的共享访问对整个化学界都很重要.寻找化学数据之间客观联系的前提是拥有足够量的数据,但化学数据并非像图像数据一样简单易得且具有良好的通用性.数据量不足很容易导致ML训练失败,无法产生所需结果.在化学合成领域中底物和催化剂的微小改变都会导致合成产物的不同,因此有效数据少之又少.当前开源化学数据集涵盖的化学类型不多、配体种类并不全面,如广泛应用于图像处理领域的ImageNet数据库[67]和涵盖诸多量子化学、物理化学信息数据的MoleculeNet数据库[68],以及收集了大量小分子化合物量化信息的GDB-17[69]及其子库QM8、QM9等,在模型训练方面均具有显著的成效.获得大量高质量数据,建立完备数据集才有可能最大限度地发挥ML在化学领域的潜力.特别是DeepChem、SchNetPack[70]等软件的发展,解决高质量数据的问题与计算化学软件包的发展息息相关.
如今,ML对反应优化、分子合成、机理探索等方面有着不容小觑的影响.但掌握影响ML结果的因素依旧很困难.在进行ML研究时,必须考虑3个关键因素[71]:数据、表示和算法.建立一套客观评价ML的标准是必要的.通过至少一组指标来评估模型,能够进一步促进ML在化学合成领域的发展,缩小ML在化学合成及表征领域的沟通代沟.例如,文献[72]中给出了较为可行的方法来比较不同工具对高质量化学数据的准确性.模型评估标准越统一,模型可解释性越强,更有利于业内外人士进行交流.
3.2 增强ML结果与实验数据的匹配
长期以来,ML模型复杂度已从线性上升到ANN.ML在化学合成领域应用更广泛是因为可以借助高通量实验或模拟研究等方式获得大量数据,辅助化学家进行产物、产率的预测,减少人力、物力的投入.然而,ML在化学合成中的应用仍然有限.虽然当前NN算法可以无限逼近非线性模型,但需要大量训练数据作为支撑,并且它能处理的数据空间有限,无法在广阔的化学空间中做到处处预测精准.一种模型有时只能针对一种特定的化学反应,在一些实际应用中并不能寻找到最佳决策.因此,增强ML模型在化学合成领域的通用性是当务之急.ML自身的可解释性不强导致研究者需要基于化学知识对模型输出结果进行解释,但有时仍会出现不具有物理意义的结果.增强模型的可解释性既可以帮助研究者更好理解模型的输出和实际意义,也能帮助研究者更快掌握模型相关信息.
在化学合成领域中,ML特别是SL一直使用黑盒方法,但黑盒方法在可解释性、通用性、可靠性方面存在缺陷.这些缺陷很有可能会限制ML的应用,有时甚至产生错误的预测结果.ML与化学实验相结合有望生成具有更好可解释性、更高预测精度、更强通用性的模型.经过化学实验验证能够及时修改训练模型中的参数设置,以期达到最佳预测结果.
3.3 描述符和ML算法的发展
欲使ML方法预测的准确性得到进一步提高,分子描述符转换时要尽可能减少有效特征损失.描述符对ML的重要性不言而喻,目前建立描述符的方式共有4种:① 使用已有的SMILES字符串、分子指纹、分子图等;② 借助Python工具包生成描述符,RDkit是常用的工具包,包含分子指纹及其相关性的计算、分子三维表示等模块;③ 使用Gaussian等量子化学软件进行DFT计算,计算分子物理化学性质,将物理化学性质进行组合生成描述符;④ 根据反应特点,自行建立描述符. 现在已有的描述符生成方式均基于化学知识生成,如Zahrt等[64]提出一种平均空间占有率的描述符,分析不同催化剂在空间中的分布,有利于后续催化剂筛选.有效的描述符能够在数据集较小的情况下获得相对较好的预测结果.未来,研究者可以改进用于获得描述符的计算方法,采取半经验方法快捷、高效地生成高质量描述符;或许还可以将化学知识与ML相结合以及将基于化学知识的模型和数据驱动模型相结合生成描述符.
在ML领域,一个基本的定理为“没有免费的午餐”.换言之,没有一种算法可以完美地解决所有问题,尤其是对于SL的算法而言,如NN算法不是在任何情况下都比RF算法有优势,反之亦然.数据集的形式或规模都会对算法产生影响,因此,科研工作者应当根据实际需求选择合适的算法,即选择正确的ML任务.不同ML算法的使用范围和应用示例如表2所示.未来,期望ML算法能够增强其通用性和可解释性.

表2 ML在化学合成及表征领域的应用
4 结语
ML强大的数据处理能力为人们提供了一条更好理解分子性质、结构的新途径,在化学领域中得到了广泛应用.在不久的将来, ML算法的快速发展无疑将扩大可用于解决典型化学任务数据处理方法的储备.目前在化学合成及表征领域,并不存在通用性好、可解释性强、精度高的模型.无论ML模型效果多么优异,它只能提供相关性,并没有因果关系.为解决上述问题,每个ML模型特别是需要借此得出结论的,均需要相关化学知识进行严格验证,确保模型没有出现过拟合等不良现象.如今,ML在化学合成及表征领域应用广泛,但如何增强模型通用性、建立模型评估标准、完备开源数据集、将ML与实验相结合以及寻找更好的描述符仍是ML在化学合成及表征领域未来发展的重大挑战.未来,ML在化学研究中的应用会持续增加,化学工作者有必要了解相关模型背后的理论框架,找到ML和化学知识之间的交叉融合点.相信在不久的未来,以ML为代表的AI技术的引入和贯通应用将对化学合成及表征领域的发展做出不可磨灭的贡献.
