甘薯苗期氮高效利用性状的GWAS分析及候选基因的筛选与验证
2023-10-23于永超范文静刘明张强强赵鹏靳容王静朱晓亚唐忠厚
于永超,范文静,刘明,张强强,赵鹏,靳容,王静,朱晓亚,唐忠厚
甘薯苗期氮高效利用性状的GWAS分析及候选基因的筛选与验证
于永超,范文静,刘明,张强强,赵鹏,靳容,王静,朱晓亚,唐忠厚
江苏徐淮地区徐州农业科学研究所/江苏徐州甘薯研究中心/农业农村部甘薯生物学与遗传育种重点实验室,江苏徐州 221131
【目的】解析甘薯氮高效利用的遗传机制,挖掘氮利用性状的关联位点及氮高效候选基因,为甘薯氮高效型分子育种、品种遗传改良提供支持。【方法】以来自世界各地的129个甘薯栽培种为材料,设置缺氮(0 mmol·L-1纯氮)和正常氮(14 mmol·L-1纯氮)处理,采用水培试验对甘薯苗期地上部生物增加量、地下部生物增加量、地上部氮累积量、地下部氮累积量、地上部氮生理利用效率和地下部氮生理利用效率共6个表型性状进行基于混合线性模型(mixed linear model,MLM)的全基因组关联分析(genome-wide association study,GWAS)。根据分析结果确定氮高效候选基因,并对候选基因进行RT-qPCR验证。【结果】6个甘薯苗期氮高效利用性状在正常氮和缺氮处理条件下存在着广泛变异。其中,缺氮处理条件下,地上部生物增加量变异系数最大,为69.5%;地下部氮生理利用效率变异系数最小,为12.1%。除地下部氮生理利用效率外,其他5个性状彼此均显著相关。GWAS定位到与地上部生物增加量、地下部生物增加量、地下部氮累积量和地上部氮生理利用效率4个性状显著关联的134个区段内888个SNP位点。筛选、过滤得到与地上部氮生理利用效率显著关联且可靠性较高的10个区段内的93个SNP标记,基因注释得到6个甘薯氮高效候选基因。RT-qPCR验证认为3个候选基因(、和)分别编码谷氨酸脱氢酶、NPH3蛋白和TIP41-like蛋白,具有进一步研究价值。【结论】在129份甘薯种质资源中共检测到888个与甘薯苗期氮高效利用性状关联的SNP位点,其中,与地上部氮生理利用效率显著相关的SNP位点93个,筛选、鉴定到6个甘薯氮高效利用的候选基因。、和存在进一步研究价值。
甘薯;氮肥利用效率;氮高效基因;全基因组关联分析;RT-qPCR
0 引言
【研究意义】甘薯((L.) Lam.)是我国重要的粮食作物,具有高产、抗逆和营养丰富的特点,随着甘薯产业技术体系的完善,研究重心逐渐向绿色高效、种质创新方向转移[1]。氮是作物生长和产量形成的基础营养元素,氮肥的合理施用直接影响甘薯源库关系的建成和薯块的生长膨大[2-3]。我国甘薯多种植在较贫瘠的沙壤地上,生产中重施氮肥容易造成肥料利用效率降低、农业污染加重和茎叶徒长等问题[4]。因此,减少氮肥施用量,挖掘氮高效基因,开展甘薯氮高效育种具有重要意义。【前人研究进展】作物氮肥利用效率(nitrogen use efficiency,NUE)是由数量性状基因座(quantitative trait locus,QTL)控制的复杂性状[5-8]。近年来,国内外通过构建双亲分离群体,利用连锁分析完成了水稻、小麦等传统作物NUE基因的挖掘研究[9-11]。然而,传统QTL定位需要构建作图群体,遗传图谱的精度很大程度取决于作图群体的大小,存在遗传背景单一、定位精度差、群体构建时间长等缺点[12-15]。全基因组关联分析(genome-wide association study,GWAS)基于等位基因间的连锁不平衡(linkage disequilibrium,LD)原理,对多个个体在全基因组范围内的遗传变异(标记)多态性进行检测,将目标性状与遗传变异进行群体水平的统计学分析,从而定位目的基因的染色体位置以及遗传效应[16-20]。GWAS分析利用广泛的自然群体,遗传变异丰富,相比连锁分析构建庞大的遗传群体,可以节约更多时间和成本[21]。Li等[22]利用230个水稻材料进行GWAS分析并结合转录组数据,成功定位到411个水稻氮高效候选基因,并确定、和3个基因作为研究方向。Liu等[23]利用GWAS定位到水稻NUE基因,该基因启动子区29 bp的InDel突变导致栽培稻在低氮条件下分蘖减少,肥料利用效率降低。甘薯是六倍体作物,染色体高度杂合,遗传背景复杂,导致甘薯数量性状的连锁分析、关联分析落后于其他作物[24-25]。随着甘薯基因组测序基本完成,为NUE等重要性状的基因定位奠定了基础。【本研究切入点】目前,甘薯氮肥养分利用的研究方向主要集中在肥料运筹对农艺性状、生理性状的影响[26-27],遗传改良研究不足。甘薯氮高效的分子机制仍不明确,缺乏对氮高效基因的挖掘与利用。【拟解决的关键问题】本研究利用129份甘薯栽培种材料,在苗期设置缺氮水培处理,将氮高效利用表型数据与重测序数据相结合进行GWAS分析,并通过RT-qPCR技术对得到的候选基因进行验证。旨在筛选出与甘薯氮利用性状显著相关的SNP位点,挖掘氮高效基因,为培育氮肥高效利用型甘薯品种提供支持。
1 材料与方法
1.1 试验材料与试验设计
选用129份来自世界各地的甘薯种质由江苏徐州甘薯研究中心(国家甘薯品种资源库)提供。试验于2021年在江苏徐州甘薯研究中心(34°16′42″N,117°17′26″E)温室内采用水培法进行。试验采用Hoagland营养液并设置2个氮水平处理:N0(0 mmol·l-1纯氮)和CK(14 mmol·l-1纯氮),其他组分一致。6月13日从苗床中剪取长势一致且无病虫害的甘薯幼苗在清水中缓苗5 d,于6月18日加入营养液,培养至20 d时进行各表型指标测定。
氮高效候选基因验证试验选择前期筛选鉴定的2个耐低氮甘薯材料(漯紫薯1号、渝紫薯6号)和2个不耐低氮甘薯材料(安平1号、徐紫薯3号)[28]。试验采用水培法进行,设置2个氮水平处理:N0(0 mmol·l-1纯氮)和CK(14 mmol·l-1纯氮)。在处理4、6和8 d对甘薯根系进行取样,液氮速冻后于-60 ℃保存,用于候选基因表达量测定。
1.2 测定项目与方法
1.2.1 氮高效利用相关指标测定 将甘薯幼苗地上部、地下部分别于105 ℃杀青30 min,80 ℃烘干至恒重,称其干重。采用全自动凯氏定氮仪(Kjeltec 8400,FOSS)测定地上部和地下部的全氮含量,计算相关指标。利用各表型性状的相对增长率下降百分比作为表型值用于GWAS分析。
地上部(地下部)生物增加量=地上部(地下部)干重-初始干重;地上部(地下部)氮累积量=地上部(地下部)氮含量×地上部(地下部)干重;地上部(地下部)氮生理利用效率=地上部(地下部)生物增加量/地上部(地下部)氮累积量;各表型性状的相对增长率下降百分比=(正常氮表型值-低氮表型值)/正常氮表型值。
1.2.2 候选基因表达量测定 氮高效候选基因包括、、、、和,提取其RNA,进行cDNA合成,并用实时荧光定量(RT-qPCR)测定基因转录水平。使用植物RNA提取试剂盒(RNApure Plant Kit,CWBIO)提取甘薯根系总RNA。使用反转录试剂盒(PrimeScrip RT reagent Kit,Takara)将提取的RNA反转录为cDNA。通过将样品cDNA与TB Green、ROX及正反引物配成RT-qPCR体系后,在QuantStudio 6 Flex Real-Time PCR System系统上进行PCR反应。每个样品设置3个生物学重复和3个技术重复。以为内参基因,采用2−ΔΔCT法[29]进行基因相对表达量分析。内参基因及相关的基因引物序列如表1所示。

表1 内参及氮高效候选基因引物序列
1.3 数据处理与分析
1.3.1 关联分析 通过对129个甘薯栽培种进行全基因组重测序,并与甘薯野生近缘种Ipomoea trifida参考基因组进行比对,经过数据质量控制过滤得到1 449 720个高质量SNP标记(江苏徐州甘薯研究中心提供)用于关联分析。利用EMMAX程序包[30]中的混合线性模型(mixed linear model,MLM),以群体结构(Q)和亲缘关系(K)为协变量加入混合线性模型中,对Q和K的效应进行评估,控制其对目的基因定位的影响。对1 449 720个SNP标记结合氮高效利用表型数据进行GWAS分析,获得甘薯氮高效候选基因。GWAS分析结果以曼哈顿图(Manhattan plot)和QQ图(Quantile-Quantile plot)展现,以-log10()≥6.16(≤1/n,n为标记个数1 449 720)为显著性阈值筛选关联位点[31]。
1.3.2 候选基因筛选及注释 根据曼哈顿图中明显峰值的位点筛选显著关联的SNP位点,将该标记物理位置两侧扩增10 kb[32-33]作为候选基因的筛选区域,再结合trifida v3参考基因组功能注释的相关数据库(http://sweetpotato.uga.edu/和https://www.ncbi.nlm. nih.gov/)进行候选基因的功能注释。
1.3.3 数据处理 使用R软件中的CMplot包进行曼哈顿图和Q-Q图绘制,使用Microsoft Excel 2016进行试验数据整理和记录,使用SPSS 25.0进行数据分析,利用Graphpad Prism 8进行基因表达量作图。
2 结果
2.1 甘薯氮高效利用表型数据分析
6个氮高效利用表型性状在正常氮和缺氮处理条件下均有广泛的变异(表2)。其中,缺氮处理条件下,地上部生物增加量变异系数最大,为69.5%,其次为地上部氮累积量,为53.9%,变异系数最小的是地下部氮生理利用效率,为12.1%。并且,除了地下部氮生理利用效率,其他性状在缺氮处理条件下的变异系数普遍升高。
通过对各表型性状进行相关性分析(表3)。地上部生物增加量、地下部生物增加量、地上部氮累积量、地下部氮累积量和地上部氮生理利用效率5个性状彼此显著相关。因此,甘薯氮高效特性较为复杂,多个性状间存在显著相关性,在多个性状中重复关联到的QTL和标记可以提高结果可靠性。
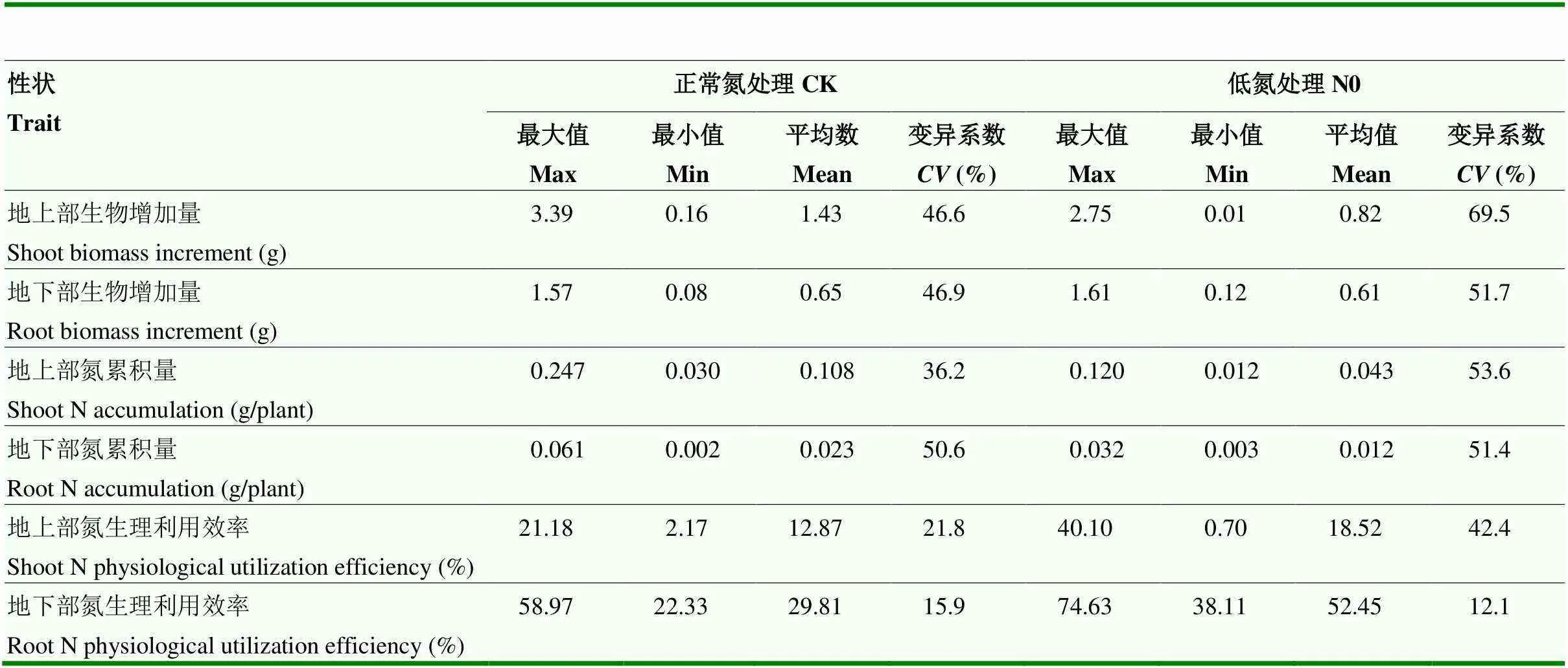
表2 氮高效利用性状的统计分析
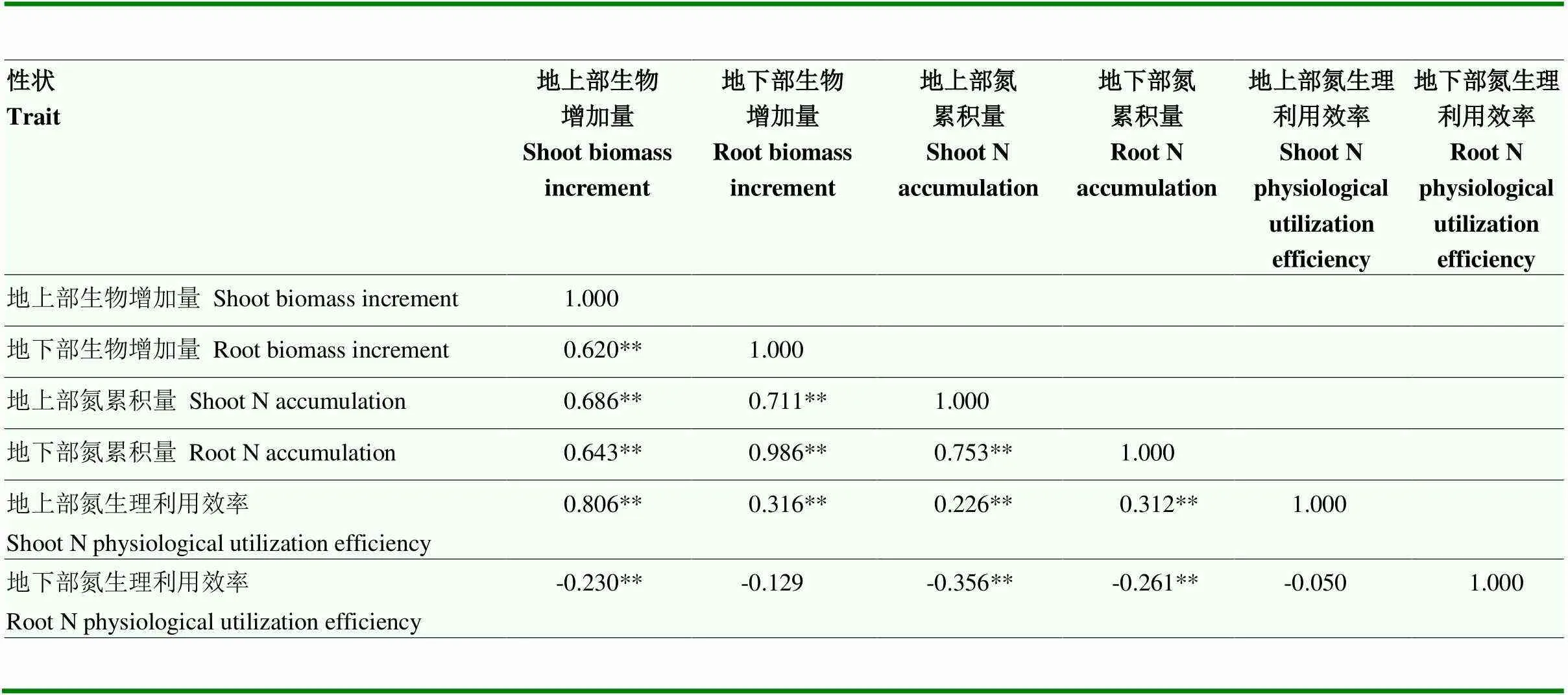
表3 氮高效利用性状的相关性分析
*和**分别代表在0.05和0.01水平差异显著 * and **indicate significant at the 0.05 and 0.01 level, respectively
2.2 甘薯氮高效利用性状的全基因组关联分析
利用MLM模型对1 449 720个SNP标记和甘薯苗期地上部生物增加量、地下部生物增加量、地上部氮累积量、地下部氮累积量、地上部氮生理利用效率、地下部氮生理利用效率6个表型性状值进行全基因组关联分析。定位到与地上部生物增加量、地下部生物增加量、地下部氮累积量和地上部氮生理利用效率4个性状显著关联的共有134个区段的888个SNP位点。其中,与地上部生物增加量显著关联的有20个区段的82个SNP位点(图1-A,图2-A);与地下部生物增加量显著关联的有36个区段的226个SNP位点(图1-B,图2-B);与地下部氮累积量显著关联的有33个区段的216个SNP位点(图1-C,图2-C);与地上部氮生理利用效率显著关联的有45个区段的364个SNP位点(图1-D,图2-D)。

A:地上部生物增加量;B:地下部生物增加量;C:地下部氮累积量;D:地上部氮生理利用效率

A:地上部生物增加量;B:地下部生物增加量;C:地下部氮累积量;D:地上部氮生理利用效率
在第1染色体22.5536—23.1333 Mb区段检测到与地上部生物增加量、地下部氮累积量和地上部氮生理利用效率3个性状稳定关联的标记位点;在第3染色体上的3个区段(7.3974—10.4446 Mb、20.9264 —21.1205 Mb、23.892063—24.931 Mb)检测到与地上部生物增加量、地下部生物增加量和地上部氮累积量稳定关联的标记位点;在第6染色体22.8417—25.6484 Mb区段检测到与地下部生物增加量和地下部氮累积量稳定关联的标记位点,地上部生物增加量和地上部氮生理利用效率性状中也有部分关联的标记位点。且从地上部氮生理利用效率性状的曼哈顿图中得出,第1染色体位置有明显峰值,第8和第13染色体位置也有部分连续峰值(图1-D),说明这些关联标记的可靠性较高。
基于曼哈顿图显著峰值,对SNP标记在多个性状中的重复关联进行筛选,剔除距离标记物理位置大于10 kb和假阳性较高的单个标记,过滤得到与地上部氮生理利用效率显著关联且可靠性较高的10个区段以及区间内的93个SNP标记(表4),分别位于第1、8和13染色体。

表4 地上部氮生理利用效率关联位点
2.3 甘薯氮高效候选基因预测
将筛选到与地上部氮生理利用效率显著关联的93个SNP标记上下游10 kb作为候选基因筛选区域,比对甘薯野生近缘种trifida参考基因组与NCBI数据库,得到92个候选基因(电子附表1)。基于功能注释预测了6个甘薯氮高效利用的候选基因。其中,基因与1_22818391标记距离4.94 kb,编码NPH3(nonphototropic hypocotyl 3)蛋白;1_22858802标记位于基因非编码区7.73 kb,编码GRP(gibberellin-regulated protein)蛋白;1_22862971、1_22863097、1_22864281、1_22870353以及1_22870858 5个标记位于候选基因的CDS区,该候选基因编码TIP41-like家族蛋白;1_23067752、1_23069513、1_23069514、1_23069538、1_23072072和1_23072074 6个标记位于的编码区,该基因编码亚硝酸还原酶(nitrite reductase,NiR);候选基因位于1_25650399标记1.49 kb,编码NAC-like转录因子;位于第1染色体的候选基因距离标记1_6780655位置大于10 kb,该候选基因编码谷氨酸脱氢酶(glutamate dehydrogenase,GDH)。
2.4 甘薯氮高效候选基因验证
通过RT-qPCR测定,耐低氮甘薯材料(漯紫薯1号、渝紫薯6号)与不耐低氮甘薯材料(安平1号、徐紫薯3号)的根系在缺氮处理下候选基因表达出现明显响应(图3)。缺氮处理后,漯紫薯1号表达水平提高,而不耐低氮的安平1号和徐紫薯3号在处理第4天、第8天则显著降低;同样,缺氮处理后漯紫薯1号表达量显著提高,徐紫薯3号明显降低,安平1号处理第8天也出现显著降低;缺氮处理后,漯紫薯1号表达水平有上升趋势,而渝紫薯6号表达量则显著降低,徐紫薯3号在处理第6天、第8天表达量也显著提高;漯紫薯1号表达量在处理第4天、第6天有所提高,而安平1号在处理第6天、第8天表达量则明显降低,徐紫薯3号在处理第4天、第8天表达量也有所降低;在4个试验材料中的表达模式不固定,没有明显规律;缺氮处理后,4个材料的表达量都发生了显著降低。
3 讨论
3.1 甘薯氮高效利用性状表型变异
我国是世界甘薯种植的第一大国,提高甘薯肥料利用效率,有助于降低环境污染,实现农业降本增效[1, 34]。苗期是甘薯氮素吸收、利用的关键时期,也是群体建成的关键期,不同基因型甘薯材料的氮素利用在苗期已经表现出差异[28]。因此,挖掘苗期氮高效基因对培育甘薯氮高效品种,提高甘薯氮肥利用效率至关重要。本研究利用129个甘薯栽培品种,在正常氮和缺氮处理下对6个氮利用表型性状(地上部生物增加量、地下部生物增加量、地上部氮累积量、地下部氮累积量、地上部氮生理利用效率和地下部氮生理利用效率)进行了平均水平和变异系数分析。研究发现,缺氮处理显著降低了甘薯地上部生物增加量、地上部氮累积量、地下部氮累积量的平均值,说明低氮胁迫会严重抑制甘薯幼苗的生长。缺氮处理条件下,甘薯的地上部氮生理利用效率和地下部氮生理利用效率得到了显著提高。因此,适量减少肥料施用同样是提高肥料利用率的有效方法。除此以外,选择的129个甘薯栽培品种氮高效利用性状变异系数较大,遗传背景丰富,容易关联到调控甘薯氮高效利用的优良候选基因[35]。
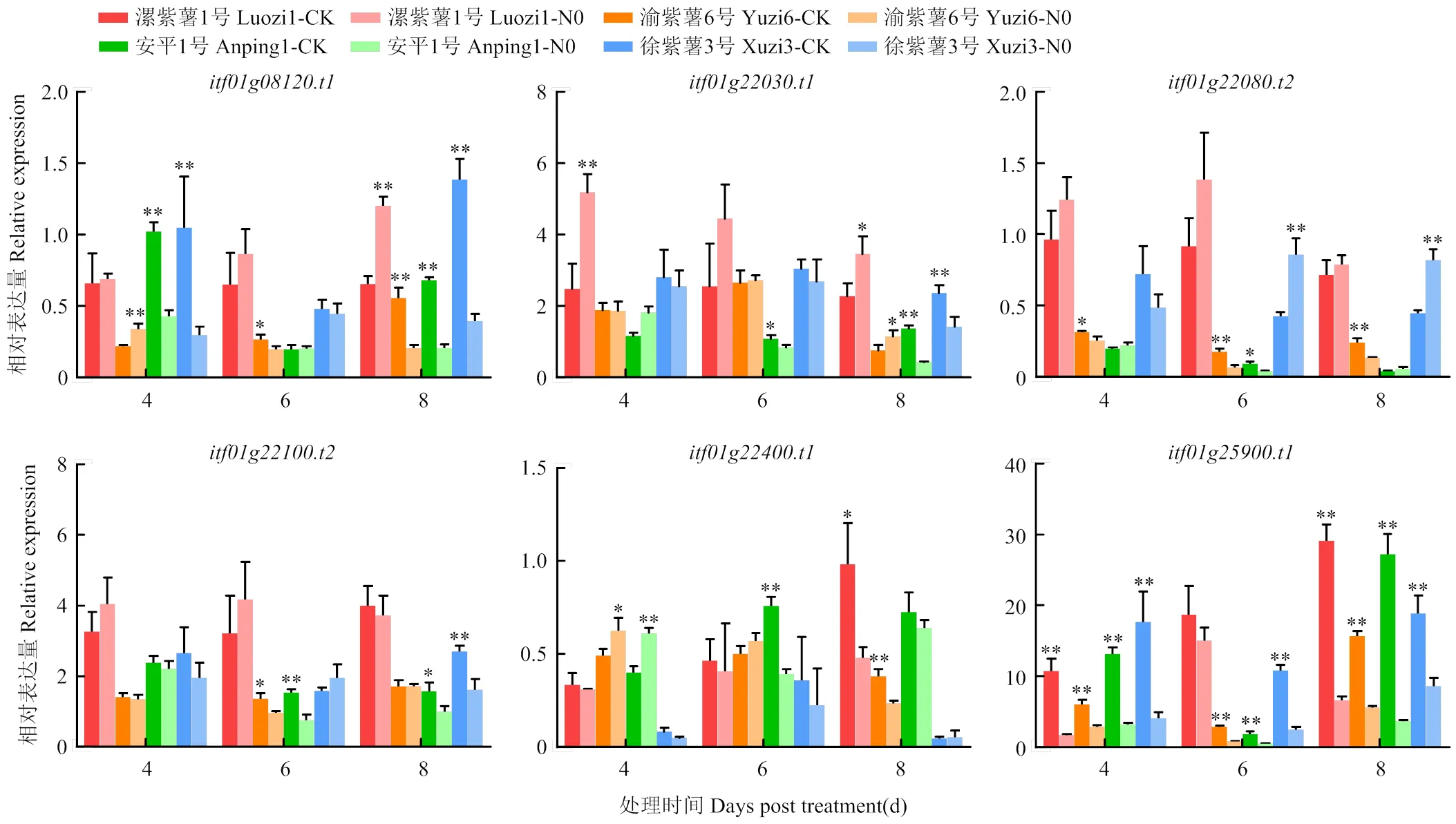
N0:缺氮处理;CK:正常氮处理。*和**分别表示在0.05和0.01水平差异显著
3.2 甘薯氮高效候选基因挖掘
利用MLM模型关联到与甘薯地上部生物增加量、地下部生物增加量、地下部氮累积量和地上部氮生理利用效率4个氮高效性状共134个区段内888个SNP标记。Q_Q图(图2)显示氮高效相关性状的期望值和观察值分离较提前,难以计算SNP位点与表型值的相关性[36]。因此,筛选在多个表型性状中稳定关联的区段和SNP位点,有助于确定候选基因。从地上部氮生理利用效率性状中筛选出的4个性状被重复检测到,可靠性较高的有93个SNP位点。其中,48个标记定位到第1染色体22.80—23.16 Mb区段;11个标记定位到第1染色体22.55—22.75 Mb区段;另有11个标记定位到第13染色体20.87—20.95 Mb区段。以上QTL区段关联到与甘薯地上部氮生理利用效率的标记较多,较大可能存在氮高效基因。
本研究以甘薯二倍体野生近缘种trifida作参考基因组,注释得到92个候选基因(电子附表1),基于研究报道和功能注释,推测出6个较大可能的甘薯氮高效候选基因并验证。其中,编码GDH,GDH可直接催化氨态氮与α-酮戊二酸合成谷氨酸,是作物谷氨酰胺合成酶(GS)/谷氨酸合成酶(GOGAT)途径之外的一种重要的氮同化途径[37-38]。目前,将低等生物高效率的GDH基因转入作物成为提高作物NUE的有效手段。Du等[39]将毛束霉属转入水稻,显著提高低氮胁迫下水稻的产量和谷蛋白、醇溶蛋白含量。Nolte等[40]也通过将大肠杆菌转入烟草,有效提高了烟草NUE,并且转基因植株表现出除草剂草铵膦抗性。和编码转录调节蛋白。编码植物中特有的NPH3家族蛋白,NPH3蛋白是植物蓝光受体向光素(phototropin,PHOT)下游信号转导因子,可与EHB1(enhanced bending1)蛋白互作调控生长素的极性运输[41]。Zhang等[42]证实,水稻通过生长素信号响应基因对下游氮代谢相关基因的激活,实现籼稻氮肥的高效利用。因此,调控生长素影响甘薯氮高效利用存在可能。编码NAC-like转录因子,NAC转录因子响应植物非生物胁迫,调控赤霉素的合成和信号转导[43]。编码赤霉素调节蛋白GRP。目前,很多研究认为赤霉素信号与作物氮肥利用存在联系。Wu等[44]通过提高水稻赤霉素信号转导途径中的关键元件GRF4(growth- regulating factor 4)和NGR5(nitrogen-mediated tiller growth response 5)的转录激活活性,在保持优良的半矮秆和高产特性下,提高水稻氮素吸收能力,在减少施氮的情况下获得更高产量。Tian等[45]也发现,编码赤霉素2-氧化酶(gibberellin 2-oxidases,GA2oxs),显著提高小麦NUE和光合速率,适量减少氮肥施用不会造成产量降低。编码TIP41-like蛋白,调控雷帕霉素靶蛋白(target of rapamycin,TOR)信号通路。Liu等[46-47]提出硝态氮和铵态氮可以激活TOR激酶进而促进拟南芥茎尖生长,推测可以影响甘薯的碳氮代谢过程。编码NiR,NiR与硝酸还原酶(nitrate reductase,NR)偶联将硝态氮还原成氨态氮进一步通过GS/GOGAT途径完成氮同化[48],可能在甘薯氮高效中发挥重要作用。
3.3 甘薯氮高效候选基因验证
前期研究筛选得到了不同氮利用类型的甘薯材料,耐低氮型甘薯材料(漯紫薯1号、渝紫薯6号)和不耐低氮型甘薯材料(安平1号、徐紫薯3号),本研究设置缺氮处理对6个氮高效候选基因进行初步验证。缺氮处理后,、和在不同氮利用类型的甘薯材料中发生了差异表达,在耐低氮的漯紫薯1号中表达量提高,不耐低氮的安平1号和徐紫薯3号中表达量降低。邱旭华[38]同样表明,的水稻同源基因在低氮处理后表达量提高,可能促进氮的利用。因此,候选基因的差异表达可能是造成不同基因型甘薯材料氮利用差异的原因。缺氮处理后,在同样是耐低氮型甘薯的漯紫薯1号和渝紫薯6号中表达模式不一致,漯紫薯1号表达量升高而渝紫薯6号显著降低,处理4 d后安平1号与徐紫薯3号的表达量也显著降低。则没有表现出明显的响应模式。因此,无法判断和在甘薯氮高效利用中的作用。缺氮处理后,漯紫薯1号、渝紫薯6号、安平1号和徐紫薯3号4个材料的表达量全部降低,推测氮源减少抑制了的转录。综上所述,候选基因、和值得深入研究。
4 结论
在129份甘薯栽培品种中共检测到888个与甘薯苗期氮高效利用性状关联的SNP位点。其中,与地上部氮生理利用效率显著相关的SNP位点93个,筛选、鉴定到6个甘薯氮高效利用的候选基因,其中,、和可进行进一步研究。
[1] 王欣, 李强, 曹清河, 马代夫. 中国甘薯产业和种业发展现状与未来展望. 中国农业科学, 2021, 54(3): 483-492.
WANG X, LI Q, CAO Q H, MA D F. Current status and future prospective of sweetpotato production and seed industry in China. Scientia Agricultura Sinica, 2021, 54(3): 483-492. (in Chinese)
[2] 唐忠厚, 李洪民, 张爱君, 史新敏, 魏猛, 陈晓光, 丁艳锋. 甘薯叶光合特性与块根主要性状对氮素供应形态的响应. 植物营养与肥料学报, 2013, 19(6): 1494-1501.
TANG Z H, LI H M, ZHANG A J, SHI X M, WEI M, CHEN X G, DING Y F. Responses of nitrogen supply forms on leaf photosynthetic characteristics and root characters of sweetpotato. Plant of Nutrition and Fertilizer Science, 2013, 19(6): 1494-1501. (in Chinese)
[3] HAN X, WU K, FU X D, LIU Q. Improving coordination of plant growth and nitrogen metabolism for sustainable agriculture. abiotech, 2020, 1(4): 255-275.
[4] 赵鹏, 刘明, 靳容, 陈晓光, 张爱君, 唐忠厚, 魏猛. 长期施用有机肥对潮土区甘薯碳氮积累与分配的影响. 中国农业科学, 2021, 54(10): 2142-2153.
ZHAO P, LIU M, JIN R, CHEN X G, ZHANG A J, TANG Z H, WEI M. Effects of long-term application of organic fertilizer on carbon and nitrogen accumulation and distribution of sweetpotato in fluvoaquic soil area.Scientia Agricultura Sinica, 2021, 54(10): 2142-2153.(in Chinese)
[5] LI P C, CHEN F J, CAI H G, LIU J C, PAN Q C, LIU Z G, GU R L, MI G H, ZHANG F S, YUAN L X. A genetic relationship between nitrogen use efficiency and seedling root traits in maize as revealed by QTL analysis. Journal of Experimental Botany, 2015, 66(11): 3175-3188.
[6] YANG X H, XIA X Z, ZHANG Z Q, NONG B X, ZENG Y, XIONG F Q, WU Y Y, GAO J, DENG G F, LI D T. QTL mapping by whole genome re-sequencing and analysis of candidate genes for nitrogen use efficiency in rice. Frontiers in Plant Science, 2017, 8: 1634.
[7] SINGH R, SARIPALLI G, KUMAR A, GAUTAM T, SINGH S K, GAHLAUT V, KUMAR S, MEHER P K, MISHRA R P, SINGH V K, SHARMA P K, BALYAN H S, GUPTA P K. QTL analysis for nitrogen use efficiency in wheat (L.). Euphytica, 2022, 219(1): 1-22.
[8] SAINI D K, CHOPRA Y, PAL N, CHAHAL A, SRIVASTAVA P, GUPTA P K. Meta-QTLs, ortho-MQTLs and candidate genes for nitrogen use efficiency and root system architecture in bread wheat (L.). Physiology and Molecular Biology of Plants, 2021, 27(10): 2245-2267.
[9] SUN H Y, QIAN Q, WU K, LUO J J, WANG S S, ZHANG C W, MA Y F, LIU Q, HUANG X Z, YUAN Q B, HAN R X, ZHAO M, DONG G J, GUO L B, ZHU X D, GOU Z H, WANG W, WU Y J, LIN H X, FU X D. Heterotrimeric G proteins regulate nitrogen-use efficiency in rice. Nature Genetics, 2014, 46(6): 652-656.
[10] WANG Q, SU Q M, NIAN J Q, ZHANG J, GUO M, DONG G J, HU J, WANG R S, WEI C S, LI G W, WANG W, GUO H S, LIN S Y, QIAN W F, XIE X Z, QIAN Q, CHEN F, ZUO J R. The Ghd7 Transcription Factor Represses theexpression to enhance nitrogen utilization and grain yield in rice. Molecular Plant, 2021, 14(6): 1012-1023.
[11] WANG Q, NIAN J Q, XIE X Z, YU H, ZHANG J, BAI J T, DONG G J, HU J, BAI B, CHEN L C, XIE Q J, FENG J, YANG X L, PENG J L, CHEN F, QIAN Q, LI J Y, ZUO J R. Genetic variations inmediate grain yield by modulating nitrogen utilization in rice. Nature Communications, 2018, 9(1): 735.
[12] 曹志斌, 李瑶, 曾博虹, 毛凌华, 蔡耀辉, 吴晓峰, 袁林峰. 非洲栽培稻垩白粒率耐热性QTL的定位. 中国水稻科学, 2020, 34(2): 135-142.
CAO Z B, LI Y, ZENG B H, MAO L H, CAI Y H, WU X F, YUAN L F.QTL mapping for heat tolerance of chalky grain rate ofsteud. Chinese Journal of Rice Science, 2020, 34(2): 135-142.(in Chinese)
[13] XU Y F, WANG R F, TONG Y P, ZHAO H T, XIE Q G, LIU D C, ZHANG A M, LI B, XU H X, AN D G. Mapping QTLs for yield and nitrogen-related traits in wheat: influence of nitrogen and phosphorus fertilization on QTL expression. Theoretical and Applied Genetics, 2014, 127(1): 59-72.
[14] LIAN X M, XING Y Z, YAN H, XU C G, LI X H, ZHANG Q F. QTLs for low nitrogen tolerance at seedling stage identified using a recombinant inbred line population derived from an elite rice hybrid. Theoretical and Applied Genetics, 2005, 112(1): 85-96.
[15] MARQUEZ-CEDILLO L A, HAYES P M, JONES B L, KLEINHOFS A, LEGGE W G, ROSSNAGEL B G, SATO K, ULLRICH S E, WESENBERG D M, NORTH AMERICAN BARLEY GENOME MAPPING PROJECT. QTL analysis of malting quality in barley based on the doubled-haploid progeny of two elite North American varieties representing different germplasm groups. Theoretical and Applied Genetics, 2000, 101: 173-184.
[16] TAM V, PATEL N, TURCOTTE M, BOSSÉ Y, PARÉ G, MEYRE D. Benefits and limitations of genome-wide association studies. Nature Reviews Genetics, 2019, 20(8): 467-484.
[17] TIBBS CORTES L, ZHANG Z W, YU J M. Status and prospects of genome‐wide association studies in plants. The Plant Genome, 2021, 14(1): e20077.
[18] WANG Q, TANG J L, HAN B, HUANG X H. Advances in genome-wide association studies of complex traits in rice. Theoretical and Applied Genetics, 2020, 133: 1415-1425.
[19] LIU C, CHEN K, ZHAO X Q, WANG X Q, SHEN C C, ZHU Y J, DAI M L, QIU X J, YANG R W, XING D Y, PANG Y, XU J L. Identification of genes for salt tolerance and yield-related traits in rice plants grown hydroponically and under saline field conditions by genome-wide association study. Rice, 2019, 12: 1-13.
[20] YOUSEF R, REZA B M, ALIREZA T, HADI A, INGVARSSON PÄR K. Genome-wide association study of agronomic traits in bread wheat reveals novel putative alleles for future breeding programs. BMC Plant Biology, 2019, 19(1): 541.
[21] SINGH B D, SINGH A K. Marker-assisted selection//Marker- assisted plant breeding: principles and practices. New Delhi: Springer India, 2015: 259-293.
[22] LI Q, LU X L, WANG C J, SHEN L, DAI L P, HE J L, YANG L, LI P Y, HONG Y F, ZHANG Q, DONG G J, HU J, ZHANG G H, REN D Y, GAO Z Y, GUO L B, QIAN Q, ZHU L, ZENG D L. Genome-wide association study and transcriptome analysis reveal new QTL and candidate genes for nitrogen‐deficiency tolerance in rice. The Crop Journal, 2022, 10(4): 942-951.
[23] LIU Y Q, WANG H R, JIANG Z M, WANG W, XU R N, WANG Q H, ZHANG Z H, LI A F, LIANG Y, OU S J, LIU X J, CAO S Y, TONG H N, WANG Y H, ZHOU F, LIAO H, HU B, CHU C C. Genomic basis of geographical adaptation to soil nitrogen in rice. Nature, 2021, 590(7847): 600-605.
[24] SASAI R M, TABUCHI H, SHIRASAWA K, KISHIMOTO K, SATO S, OKADA Y, KURAMOTO A, KOBAYASHI A, ISOBE S, TAHARA M, MONDEN Y. Development of molecular markers associated with resistance to meloidogyne incognita by performing quantitative trait locus analysis and genome-wide association study in sweetpotato. DNA Research, 2019, 26(5): 399-409.
[25] 马猛. 紫甘薯SSR标记遗传图谱构建及主要农艺性状QTL定位[D]. 北京: 中国农业科学院, 2021.
MA M.Construction of SSR marker genetic map and QTL mapping of main agronomic traits in purple sweet potato[D]. Beijing: Chinese Academy of Agricultural Sciences, 2021. (in Chinese)
[26] 宁运旺, 马洪波, 张辉, 汪吉东, 许仙菊, 张永春. 甘薯源库关系建立、发展和平衡对氮肥用量的响应. 作物学报, 2015, 41(3): 432-439.
NING Y W, MA H B, ZHANG H, WANG J D, XU X J, ZHANG Y C. Response of sweetpotato in source-sink relationship establishment, expanding, and balance to nitrogen application rates. Acta Agronomica Sinica, 2015, 41(3): 432-439. (in Chinese)
[27] 刘明, 李洪民, 张爱君, 陈晓光, 靳容, 蒋薇, 唐忠厚. 不同氮肥与密度水平对鲜食甘薯产量和品质的影响. 华北农学报, 2020, 35(1): 122-130.
LIU M, LI H M, ZHANG A J, CHEN X G, JIN R, JIANG W, TANG Z H. Effects of nitrogen fertilizer and density on yield and quality of fresh edible type sweetpotato. Acta Agriculturae Boreali-Sinica, 2020, 35(1): 122-130. (in Chinese)
[28] 范文静, 刘明, 赵鹏, 张强强, 吴德祥, 郭鹏宇, 朱晓亚, 靳容, 张爱君, 唐忠厚. 甘薯苗期耐低氮基因型筛选及不同氮效率类型综合评价. 中国农业科学, 2022, 55(10): 1891-1902.
FAN W J, LIU M, ZHAO P, ZHANG Q Q, WU D X, GUO P Y, ZHU X Y, JIN R, ZHANG A J, TANG Z H.Screening of sweetpotato varieties tolerant to low nitrogen at seedling stage and evaluation of different nitrogen efficiencies.Scientia Agricultura Sinica,2022, 55(10): 1891-1902. (in Chinese)
[29] LIVAK K J, SCHMITTGEN T D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCTmethod. Methods, 2001, 25(4): 402-408.
[30] KANG H M, SUL J H, SERVICE S K, ZAITLEN N A, KONG S Y, FREIMER N B, SABATTI C, ESKIN E. Variance component model to account for sample structure in genome-wide association studies. Nature Genetics, 2010, 42(4): 348-354.
[31] LI Y, CAO K, ZHU G R,FANG W C, CHEN C W, WANG X W, ZHAO P, GUO J, DING T Y, GUAN L P, ZHANG Q, GUO W W, FEI Z J, WANG L R. Genomic analyses of an extensive collection of wild and cultivated accessions provide new insights into peach breeding history. Genome Biology, 2019, 20(1): 36.
[32] 贵会平, 董强, 张恒恒, 王香茹, 庞念厂, 王准, 刘记, 郑苍松, 付小琼, 张西岭, 宋美珍. 棉花苗期耐低氮基因型初步筛选.棉花学报, 2018, 30(4): 326-337.
GUI H P, DONG Q, ZHANG H H, WANG X R, PANG N C, WANG Z, LIU J, ZHENG C S, FU X Q, ZHANG X L, SONG M Z.Preliminary screening of low nitrogen-tolerant cotton genotypes at seedling stage. Cotton Science, 2018, 30(4): 326-337. (in Chinese)
[33] 房增国, 高璐阳. 8个鲜食型甘薯品种的氮营养差异研究. 作物杂志, 2015(1): 86-90.
FANG Z G, GAO L Y.Differences of nitrogen nutrition of eight fresh-eating sweet potato cultivars. Crops, 2015(1): 86-90. (in Chinese)
[34] 李强, 赵海, 靳艳玲, 朱金城, 马代夫. 中国甘薯产业助力国家粮食安全的分析与展望. 江苏农业学报, 2022, 38(6): 1484-1491.
LI Q, ZHAO H, JIN Y L, ZHU J C, MA D F.Analysis and perspectives of sweetpotato industry contributing to national food security in China. Jiangsu Journal of Agricultural Sciences, 2022, 38(6): 1484-1491. (in Chinese)
[35] 严勇亮, 张恒, 张金波, 时晓磊, 耿洪伟, 肖菁, 路子峰, 倪中福, 丛花. 春小麦主要籽粒性状的全基因组关联分析. 麦类作物学报, 2022, 42(10): 1182-1191.
YAN Y L, ZHANG H, ZHANG J B, SHI X L, GENG H W, XIAO J, LU Z F, NI Z F, CONG H.Genome-wide association study of grain traits of spring wheat. Journal of Triticeae Crops, 2022, 42(10): 1182-1191. (in Chinese)
[36] WANG S B, FENG J Y, REN W L, HUANG B, ZHOU L, WEN Y J, ZHANG J, DUNWELL J M, XU S Z, ZHANG Y M. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Scientific Reports, 2016, 6(1):19444.
[37] KWINTA J, BIELAWSKI W. Glutamate dehydrogenase in higher plants. Acta Physiologiae Plantarum, 1998, 20(4):453-463.
[38] 邱旭华. 水稻氮代谢基础研究: 谷氨酸脱氢酶作用的分子机理[D]. 武汉: 华中农业大学, 2009.
QIU X H. Basic research on nitrogen metabolism in rice: molecular mechanism of glutamate dehydrogenase[D]. Wuhan: Huazhong Agricultural University, 2009. (in Chinese)
[39] DU C Q, LIN J Z, DONG L A, LIU C, TANG D Y, YAN L, CHEN M D, LIU S, LIU X M. Overexpression of an NADP (H)-dependent glutamate dehydrogenase gene,, from Trichurus improves nitrogen assimilation, growth status and grain weight per plant in rice. Breeding Science, 2019, 69(3): 429-438.
[40] NOLTE S A, YOUNG B G, MUNGUR R, LIGHTFOOT D A. The glutamate dehydrogenase geneincreased the resistance of tobacco to glufosinate. Weed Research, 2004, 44(4): 335-339.
[41] 乔新荣, 陈琼. 植物向光素信号通路中NPH3蛋白的研究进展. 植物生理学报, 2015, 51(6): 829-834.
QIAO X R, CHEN Q.Advances of NPH3in plant phototropin signaling. Plant Physiology Communications, 2015, 51(6): 829-834. (in Chinese)
[42] ZHANG S Y, ZHU L M, SHEN C B, JI Z, ZHANG H P, ZHANG T, LI Y, YU J P, YANG N, HE Y B, TIAN Y N, WU K, WU J Y, HARBERD N P, ZHAO Y D, FU X D, WANG S K, LI S. Natural allelic variation in a modulator of auxin homeostasis improves grain yield and nitrogen use efficiency in rice. The Plant Cell, 2021, 33(3): 566-580.
[43] Chen H I, Li P F, Yang C H. NAC-like generegulates the gibberellin metabolic pathway in response to cold and drought stresses in. Scientific Reports, 2019, 9: 19226.
[44] WU K, WANG S N, SONG W Z, ZHANG J Q, WANG Y, LIU Q, YU J P, YE Y F, LI S, CHEN J F, ZHAO Y, WANG J, WU X K, WANG M Y, ZHANG Y J, LIU B M, WU Y J, HARBERD N P, FU X D. Enhanced sustainable green revolution yield via nitrogen- responsive chromatin modulation in rice. Science, 2020, 367(6478): eaaz2046.
[45] Tian X L, Xia X C, Xu D G, Liu Y Q, Xie L, Hassan M A, Song J, Li F J, Wang D S, Zhang Y, Hao Y F, Li G Y, Chu C C, He Z H, Cao S H., an ancient variation of, reduces plant height without yield penalty in wheat. New Phytologist, 2022, 233(2): 738-750.
[46] BECK T, HALL M N. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature, 1999, 402(6762): 689-692.
[47] LIU Y L, DUAN X L, ZHAO X D, DING W L, WANG Y W, XIONG Y. Diverse nitrogen signals activate convergent ROP2-TOR signaling in. Developmental Cell, 2021, 56(9): 1283-1295.
[48] 梁喜欢, 龚丝雨, 尼金玉, 杨帅强, 王子璇, 王俊, 刘齐元. 不同基因型烟草氮代谢关键酶活性对低氮胁迫的响应. 分子植物育种, 2020, 18(22): 7554-7561.
LIANG X H, GONG S Y, NI J Y, YANG S Q, WANG Z X, WANG J, LIU Q Y.Response of key enzymes activity of nitrogen metabolism to low nitrogen stress in different genotypes tobacco. Molecular Plant Breeding, 2020, 18(22): 7554-7561. (in Chinese)
Genome-Wide Association Study of Nitrogen Use Efficient Traits in Sweetpotato Seeding Stage and Screening and Validation of Candidate Genes
YU Yongchao, FAN Wenjing, LIU Ming, ZHANG Qiangqiang, ZHAO Peng, JIN Rong, WANG Jing, ZHU Xiaoya, TANG Zhonghou
Xuzhou Institute of Agricultural Sciences of Xuhuai District of Jiangsu Province/Xuzhou Sweetpotato Research Center of Jiangsu Province/Key Laboratory of Sweetpotato Biology and Genetic Breeding, Ministry of Agriculture and Rural Affairs, Xuzhou 221131, Jiangsu
【Objective】The objective of this paper was to analyze the genetic mechanisms of nitrogen use efficiency (NUE), and to explore the loci and candidate genes associated nitrogen (N) efficient traits, to provide support for the N-efficient molecular breeding and genetic improvement of sweetpotato.【Method】A total of 129 sweetpotato cultivars from all over the world were treated with N deficiency (0 mmol·L-1) and normal N (14 mmol·L-1). A hydroponic experiment was conducted to facilitate the genome-wide association study (GWAS) of six phenotypic traits (shoot biomass increment, root biomass increment, shoot N accumulation, root N accumulation, shoot N physiological utilization efficiency, and root N physiological utilization efficiency) of sweetpotato at the seedling stage. The N-efficient candidate genes were identified based on the GWAS and subsequently- verified using RT-qPCR.【Result】There were wide variations among the six traits related to NUE in sweetpotato under the normal N and N deficiency treatment conditions. The coefficient of variation () of the shoot biomass increment under the N deficiency treatment condition was the greatest at 69.5%. The CV of the root N physiological utilization efficiency under N deficiency treatment condition was the smallest at 12.1%. All five traits were significantly correlated except for root N physiological utilization efficiency. The MLM model was used to conduct a GWAS of the six phenotypic trait values. A total of 134 QTL and 888 SNP loci were identified as being significantly associated with four out of the six traits, namely, shoot biomass increment, root biomass increment, root N accumulation, and shoot N physiological utilization efficiency. A total of 93 SNP markers across ten regions were significantly associated with shoot N physiological utilization efficiency with a high reliability. Six N efficiency candidate genes were obtained via gene annotation. RT-qPCR verified that the three candidate genes (,and) encoded glutamate dehydrogenase, NPH3 protein and TIP41-like protein, respectively, which warrants further research.【Conclusion】A total of 888 SNP loci associated with N utilization traits were detected in 129 sweetpotato cultivars. Among these, 93 SNP loci were significantly associated with shoot N physiological utilization efficiency, and six candidate genes were identified. Preliminary verification indicated that the,andgenes hold promising value for further research.
sweetpotato; nitrogen use efficiency; nitrogen efficient gene; GWAS; RT-qPCR
10.3864/j.issn.0578-1752.2023.18.002
2023-04-03;
2023-05-08
国家甘薯产业技术体系建设项目(CARS-10)
于永超,E-mail:yychaomail@163.com。范文静,E-mail:1394441094@qq.com。于永超和范文静为同等贡献作者。通信作者唐忠厚,E-mail:zhonghoutang@sina.com
(责任编辑 李莉)
