非洲猪瘟病毒多重PMA-qPCR检测方法的建立及应用
2023-10-19卓玛钱炳旭吴晓东戴建君薛峰
卓玛,钱炳旭,吴晓东,戴建君,薛峰*
(1. 教育部动物卫生与食品安全国际联合研究实验室,江苏 南京 210000;2. 南京农业大学三亚学院,海南 三亚 572000;3. 中国动物卫生与流行病学中心,山东 青岛 266000)
非洲猪瘟(African swine fever,ASF)是一种急性、高度传染性的猪病,在世界范围内导致大量猪死亡,给全球养猪业带来巨大经济损失[1]。非洲猪瘟于1921年在肯尼亚被首次确诊,1957至1972年传入欧洲、美洲;2007年传入欧亚接壤的格鲁吉亚,随后传入俄罗斯并在高加索地区安家[2];2012至2018年传入乌克兰、白俄罗斯、欧盟立陶宛、匈牙利等国家[3];2018年8月以来在中国多地陆续发现疫情[4]。
由于目前尚无有效的非洲猪瘟疫苗[5-7],因此非洲猪瘟的防控主要依靠严格的生物安全措施。已有研究显示,CD2v和MGF是非洲猪瘟病毒(ASFV)2个毒力基因,敲除后ASFV的毒力显著降低[8-10],常用于作为ASFV减毒活疫苗缺失的靶点。目前,可鉴别检测ASFV野毒株与基因缺失株的方法研究相对较少。为此,本研究选择ASFV p72、CD2v、MGF110-14L基因,拟建立一种可鉴别ASFV野毒株与常见的疫苗候选缺失株的多重荧光定量PCR(qPCR)检测方法,为ASFV的日常检测提供便捷。当前,我国针对ASFV的检测依旧以OIE推荐的普通PCR和荧光定量PCR为主[11-14]。赵凯颖等[15]建立了ASFV实时荧光RAA检测方法。但是,普通PCR或荧光定量PCR检测方法存在一个明显缺陷,即只能定性检测病毒核酸,而不能有效鉴别样品中是否具有感染性的活病毒。也就是说,只要样品中存在一定量的病毒,不管是完整的具有感染性的活病毒还是通过加热、紫外线等手段破外了遗传物质但保留了病毒核酸的灭活病毒,当使用普通PCR或荧光定量PCR检测时均会对其进行扩增。因此,迫切需要一种可以鉴别ASFV活/死病毒的鉴别检测方法。准确鉴别临床样品或饲料中是否存在具有感染性的活病毒,对非洲猪瘟的防控具有重要意义。随着科技的不断发展,出现了很多可鉴别检测活/死微生物的检测方法,其中包括流式细胞分选技术、拉曼光谱检测方法、病毒分离培养等方法。董振华等[16]利用分离培养与拉曼光谱技术相结合,监测区分沙门菌为死菌还是活菌;Schaad[17]利用Bio-PCR方法达到检测活菌的目的;Lin等[18]通过脱氧胆酸钠-单叠氮丙啶-qPCR快速准确地检测虾中的活副溶血性弧菌。
叠氮溴化丙锭(PMA)是一种新型的核酸结合染料,能够穿过细胞膜不完整的细胞,经过暗孵育以及曝光处理后,PMA与DNA结合形成稳定的共价键,从而修饰细胞死亡后暴露出来的DNA分子[19-21]。通常将PMA与qPCR结合,可同时达到定量和精确检测感染性病毒与非感染性病毒(活/死病毒)核酸的目的。PMA在食品以及植物病原菌检测活菌研究中已被广泛应用,如于璇等[22]利用PMA与qPCR技术相结合,建立了十字花科黑斑病菌(Pseudomonassyringaepv.maculicola)活菌的检测方法。本研究将PMA与qPCR技术相结合,建立了一种ASFV多重PMA-qPCR检测方法,既可实现单管鉴别ASFV野毒株与基因缺失株又可以同时鉴别检测ASFV活/死病毒,为临床检测ASFV带来更多的便捷。
1 材料与方法
1.1 病毒株、载体和样本
猪肺泡巨噬细胞(PAMs)、ASFV、猪细小病毒(PPV)、猪繁殖与呼吸综合征病毒(PRRSV)、伪狂犬病毒(PRV)、猪流行腹泻病毒(PEDV)、口蹄疫病毒(FMDV)、猪德尔塔冠状病毒(PDCoV)、猪圆环病毒2型(PCV2)、塞内卡病毒(SVA)、pET-28a载体和23份临床阳性样品,均由中国动物卫生流行病学中心提供。
1.2 引物、探针的设计与合成
根据ASFV China/2018/Anhui毒株序列,利用Bio Edit软件分析核酸序列,应用Snape Gene针对ASFV p72、CD2v、MGF1110-14L基因的保守序列分别设计特异性引物与探针(表1)。引物和探针均由通用(安徽)生物技术有限公司合成并纯化。

表1 引物与探针序列

表2 多重PMA-qPCR与普通qPCR方法检测实际样品
1.3 ASFV多重qPCR方法的建立
1.3.1 重组质粒标准品的构建与鉴定
ASFV China/2018/Anhui毒株由中国动物卫生与流行病学中心提供并保存,利用病毒核酸提取试剂盒提取ASFV基因组DNA并以其为模板,利用普通PCR扩增目的基因片段。PCR反应体系:Green Mix 12.5 μL;上下游引物各1 μL;ASFV DNA 2 μL;去离子水补充至25 μL总体积。反应程序:94 ℃ 5 min;94 ℃ 30 s,52 ℃ 30 s,72 ℃ 20 s,35个循环;72 ℃ 10 min。
将ASFV(p72、CD2v、MGF110-14L)基因的PCR产物与pClone007载体连接、转化至DH5ɑ感受态细胞,氨苄固体培养基上过夜培养,菌液PCR鉴定正确后送至擎科(南京)生物技术有限公司测序,命名为pClone007- p72、pClone007-CD2v、pClone007-MGF110-14L。提取质粒,利用酶标仪测定质粒浓度,计算拷贝数。 去离子水将质粒标准品进行10倍连续稀释,稀释至质粒终浓度为1×1012~1×101copies/μL,-20 ℃保存备用。
1.3.2 多重qPCR引物、探针、退火温度优化
采用方阵法优化引物、探针浓度,总体积为25 mL,其中2×PerfectStart Ⅱ Probe qPCR Super Mix 10 μL,引物F和R(10 μmol/L)各自加0.4、0.6、0.8、1.0、1.2 μL,探针P(10 μmol/L)加0.4、0.5、0.6、0.7、0.8 μL,Pclone007- p72、Pclone007-CD2v、Pclone007-MGF110-14L重组质粒标准品各2 μL,混匀后作为模板,去离子水补充至25 μL。使用优化好的引物和探针体系优化退火温度(56~64 ℃)。
1.3.3 特异性试验
ASFV pClone007- p72、pClone007-CD2v、pClone007-MGF110-14L、PPV、PRRSV、PRV、CSFV、PEDV、FMDV、PDCoV、PCV-2、SVA病毒核酸为模板,去离子水为阴性对照,按优化好的反应体系进行多重qPCR扩增,以验证该方法的特异性。
1.3.4 灵敏度试验
分别以10倍连续稀释的(102~108copies/μL)pClone007-p72、pClone007-CD2v、pClone007-MGF110-14L质粒标准品混合液作为模板,去离子水为阴性对照,Ct值≥35判定为阴性,进行多重qPCR扩增,以质粒标准品稀释倍数为横坐标,Ct值为纵坐标,验证该方法的最低检出限。
1.4 PMA处理条件的优化
1.4.1 活/灭活病毒悬液制备
将ASFV感染PAMs细胞后,收集上清液,抽提病毒DNA,计算拷贝数。将病毒原液制备成1×106copies/μL,制得ASFV病毒悬液,另取1×106copies/μL ASFV病毒悬液在100 ℃下灭活15 min后制得ASFV灭活病毒悬液。
1.4.2 抑制灭活病毒扩增不抑制活病毒扩增的最适PMA浓度优化
各取1×106copies/μL ASFV活/灭活病毒悬液,分别加入不同质量PMA使其终质量浓度为0、5、10、15、20、25 μg/mL,避光孵育15 min(后续进行优化),利用卤素灯曝光15 min(后续进行优化),光照强度设为60 W(后续进行优化),提取DNA后进行多重PMA-qPCR反应。
1.4.3 暗孵育时间优化
选取1×106copies/μL ASFV灭活病毒悬液,取6份200 μL灭活病毒悬液加入PMA混匀使其终质量浓度为10 μg/mL,避光孵育0、5、10、15、20、25 min,曝光15 min(后续继续优化),光照强度60 W,提取DNA后进行多重PMA-qPCR反应。
1.4.4 光照时间优化
选取1×106copies/μL ASFV灭活病毒悬液,取6份200 μL灭活病毒悬液,加入PMA混匀使其终质量浓度为10 μg/mL,避光孵育10 min,光照0、5、10、15、20、25 min,光照强度60 W (后续优化),提取DNA后进行多重PMA-qPCR反应。
1.4.5 光照强度优化
选取1×106copies/μL ASFV灭活病毒悬液,取6份200 μL灭活病毒悬液加入PMA混匀使其终浓度10 μg/mL,避光孵育10 min,曝光15 min,光照强度0、20、40、60、80、100 W,提取DNA后进行多重PMA-qPCR反应。
1.5 多重PMA-qPCR的灵敏度与普通qPCR对比
取10倍梯度连续稀释的活病毒悬液(101~108copies/μL)各200 μL,按优化好的PMA条件处理,提取DNA后进行多重PMA-qPCR反应,另设不加PMA对照组,即普通qPCR反应,以病毒稀释倍数为横坐标,Ct值为纵坐标。Ct值小于35即可判定为阳性。
1.6 多重PMA-qPCR方法检测人工感染样品
为验证PMA对ASFV非感染性病毒扩增产生抑制效果,将1.4.1方法中制备的1×106copies/μL ASFV活/灭活病毒悬液,各取1 mL加入4份ASFVp72基因临床阴性样品(Ct值=42)中,经PMA处理后进行多重PMA-qPCR检测,分别验证人工感染的ASFV活/灭活病毒样品中活病毒存活数量,同时采用普通qPCR反应进行对比。
1.7 多重PMA-qPCR方法检测实际样品
以中国动物卫生与流行病学中心提供的23份阳性临床样本,各取200 μL制备病毒悬液,利用OIE推荐的普通qPCR方法与本研究建立的多重PMA-qPCR检测方法,分别对23份临床样本进行验证。同时,将这23份样品100 ℃灭活15 min后按照上述两种方法分别检测,并比较其检测结果,计算多重PMA-qPCR方法检测活病毒的准确率。
2 结果与分析
2.1 多重qPCR检测方法建立
2.1.1 重组质粒标准品的构建与鉴定
使用ASFV DNA为模板,利用设计好的引物进行PCR扩增,结果显示,扩增后获得与预期大小一致的目的基因片段,依次为p72(158 bp)、CD2v(124 bp)、MGF110-14L(156 bp)。将目的片段回收纯化后克隆于pClone007载体上,构建重组质粒pClone007- p72、pClone007-CD2v、pClone007-MGF110-14L,重组质粒经菌落PCR鉴定和测序鉴定正确,测得重组质粒浓度分别为193、172.4和200 ng/μL,换算成拷贝数分别为11.4×1011、13.2×1011和3×1011copies/mL,作为多重qPCR的标准品。
2.1.2 多重荧光定量PCR引物、探针、退火温度优化
经优化后,确定反应体系为25 μL:2×PerfectStart II Probe qPCR Super Mix 10 μL;引物浓度分别为p72 F和R(10 μmol/L)各加0.4 μL,p72 P(10 μmol/L)加0.2 μL,CD2v F和R(10 μmol/L)各加0.4 μL,CD2v P(10 μmol/L)加0.3 μL,MGF110-14L F和R(10 μmol/L)各加0.3 μL,MGF110-14L P(10 μmol/L)加0.6 μL;3个基因的质粒标准品各3 μL混匀后作为模板;去离子水补充至25 μL总体积。扩增温度为60 ℃,整个扩增包括45个循环时qPCR具有良好的扩增曲线。
2.1.3 特异性试验
按照1.3.3验证该方法的特异性,结果显示,该方法对ASFV- p72、ASFV-CD2v、ASFV-MGF110-14L重组质粒标准品的扩增结果为阳性,其余结果均为阴性(图1)。表明本研究建立的方法具有较强的特异性。
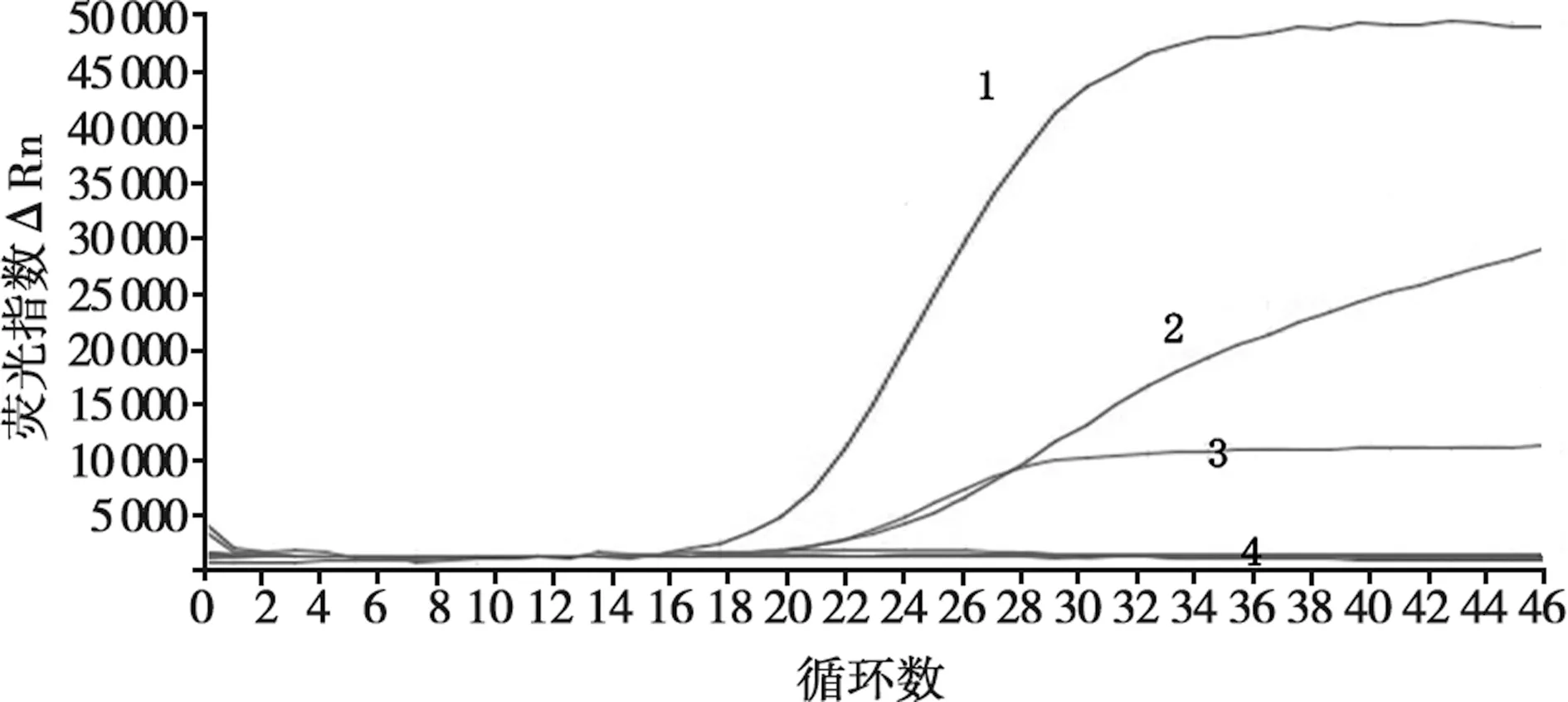
1. p72基因;2. CD2v基因;3. MGF110-14L基因;4. 阴性对照和PPV、PRRSV、PRV、CSFV、PEDV、FMDV、PDCoV、PCV-2、SVA病毒核酸
2.1.4 敏感性试验
将pClone007- p72、pClone007-CD2v、pClone007-MGF110-14L3个重组质粒标准品原液制备成3×108copies/mL,各取10 μL混匀后连续稀释,Ct值≥35为阴性。结果显示,多重qPCR方法的最低检测线为102copies/mL(图2)。表明本研究建立的多重qPCR检测方法有较高的灵敏性。

A:1~8. 101~108copies/mL的Pclone007-p72重组质粒标准品;N.阴性对照;B:1~8. 101~108copies/mL的Pclone007-CD2v重组质粒标准品;N.阴性对照; C:1~8. 101~108copies/mL的Pclone007-MGF110-14L重组质粒标准品;N.阴性对照
2.2 PMA处理条件的优化
2.2.1 PMA处理浓度优化结果
选取1.4.1方法中制备好的1×106copies/μL ASFV活/灭活病毒悬液,各取200 μL活/灭活病毒悬液分别加入PMA终浓度为0、5、10、15、20、25 μg/mL,提取DNA后进行多重PMA-qPCR扩增。图3A结果所示,活病毒悬液中随着PMA质量浓度的增大Ct值稍有波动但不明显,当加入PMA终质量浓度为10 μg/mL时,不抑制活病毒的扩增。图3B结果所示,灭活病毒悬液中随着PMA质量浓度的增大,Ct值也逐渐上升,抑制扩增效果越明显,当加入PMA终质量浓度为10 μg/mL时,Ct值趋于平衡,继续增加PMA终质量浓度对Ct值的影响不大。因此,选择PMA终质量浓度10 μg/mL为本研究最适PMA处理浓度。
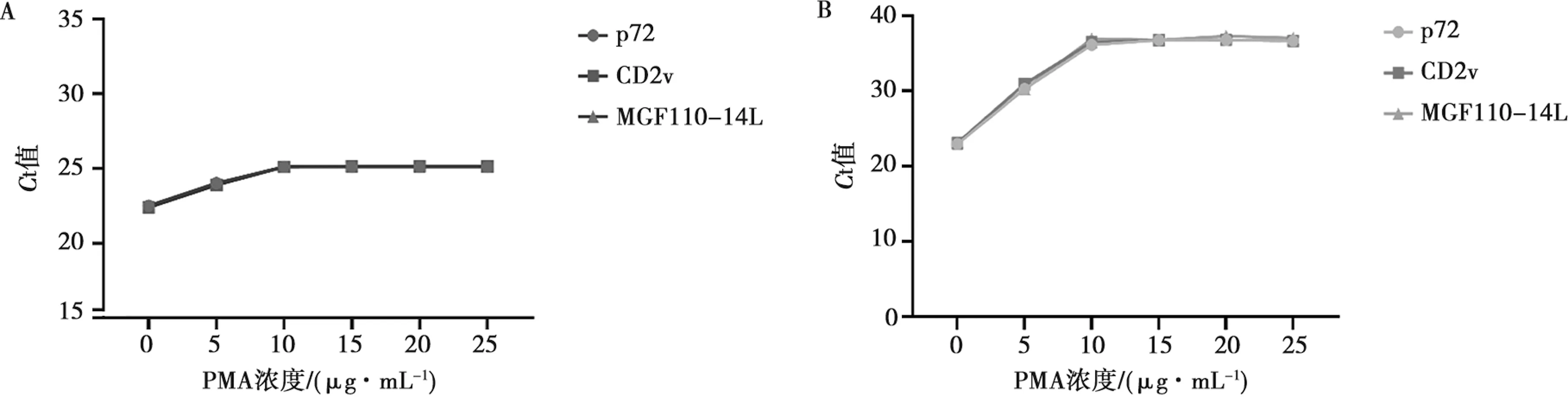
A.活病毒悬液添加不同PMA浓度;B.死病毒悬液添加不同PMA浓度

A.PMA暗孵育时间优化;B.PMA光照时间优化;C.PMA曝光强度优化
A. p72基因(PMA-qPCR);B. CD2v基因(PMA-qPCR);C. MGF110-14L基因(PMA-qPCR);D. p72基因(普通qPCR)

A. 普通qPCR检测4份人工感染的活病毒样品;B. 多重PMA-qPCR 检测4份人工感染的活病毒样品;C. 普通qPCR检测4份人工感染的灭活病毒样品;D. 多重PMA-qPCR检测4份人工感染的灭活病毒样品
2.2.2 暗孵育时间优化
取1×106copies/μL ASFV灭活病毒悬液200 μL,加入PMA混匀使其终质量浓度为10 μg/mL,避光孵育0、5、10、15、20、25 min,光照强度60 W,光照15 min,进行多重PMA-qPCR。结果显示,当避光孵育10 min时,Ct值达到37,继续延长时间对Ct值的影响不大(图4A)。因此,选择最适的暗孵育时间为10 min。
2.2.3 光照时间优化
取1×106copies/μL ASFV灭活病毒悬液200 μL,加入PMA混匀使其终质量浓度为10 μg/mL,避光孵育10 min,光照时间0、5、10、15、20、25 min,光照强度60 W,进行多重PMA-qPCR。结果显示,当光照15 min时,Ct值为37,随着光照时间的延长Ct值并无明显变化(图4B)。为节省试验时间不必继续延长试验时间,因此选择最理想的曝光时间为15 min。
2.2.4 光照强度优化
取6份1×106copies/μL ASFV灭活病毒悬液200 μL,加入PMA使其终质量浓度为10 μg/mL,避光孵育10 min,曝光15 min,光照强度0、20、40、60、80、100 W,进行多重PMA-qPCR。结果显示,当卤素灯光照强度为40 W时,达到抑制ASFV灭活病毒悬液扩增的理想条件(图4C)。
通过对PMA浓度、暗孵育时间、曝光时间以及曝光强度进行优化后,确定了PMA终质量浓度10 μg/mL、暗孵育10 min、光照强度40 W、光照15 min时PMA实现对ASFV活病毒扩增不受抑制,同时抑制灭活病毒扩增的多重PMA-qPCR理想效果。
2.3 多重PMA-qPCR的灵敏度
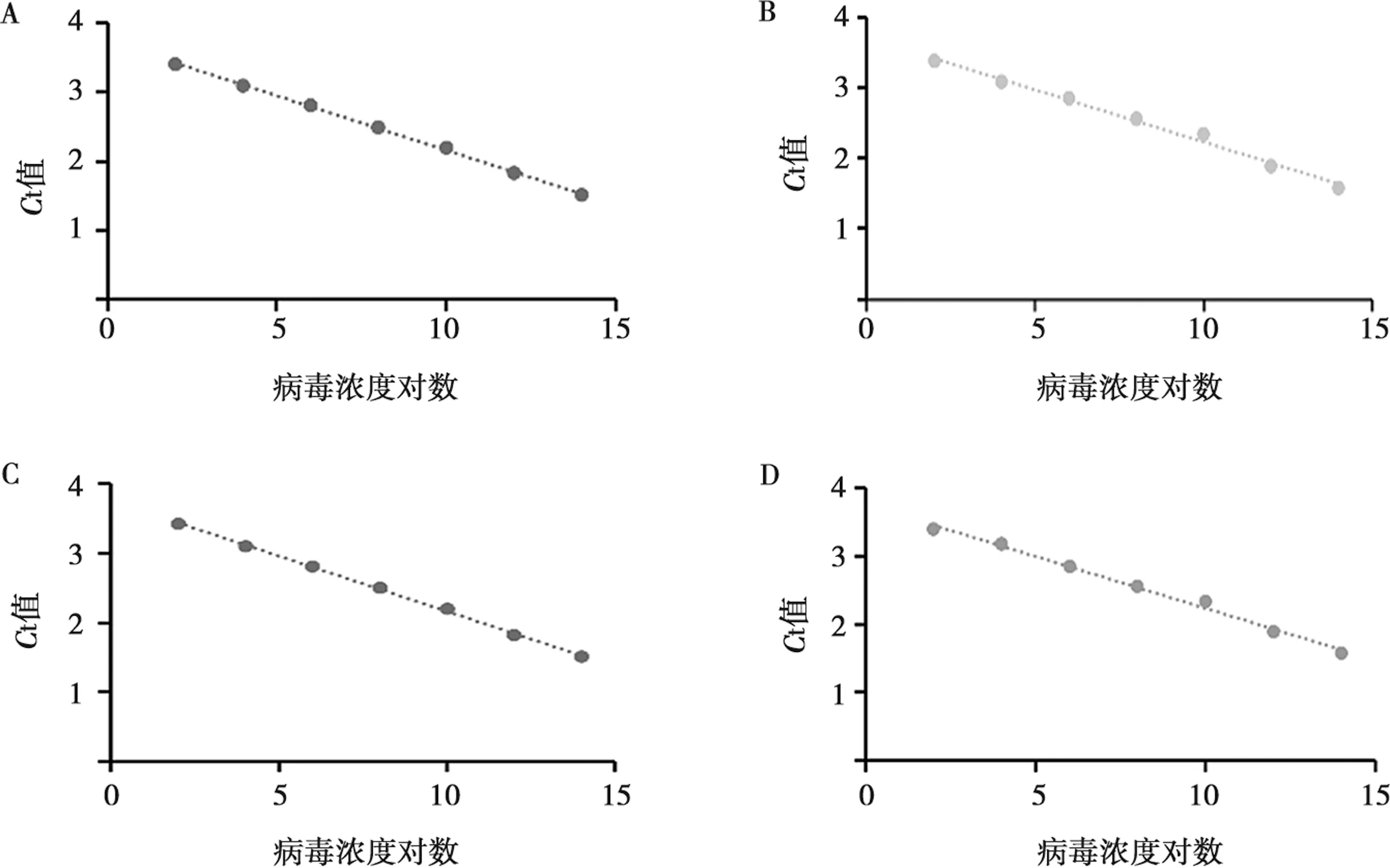
为验证该方法的灵敏性,本研究利用多重PMA-qPCR与普通qPCR方法进行灵敏度对比。结果显示,多重PMA-qPCR和普通qPCR方法的灵敏度均为102copies/μL。
多重PMA-qPCR。p72:y=3.158x+3.742 7,R2=0.999(图5A);CD2v:y=0.148x+3.71,R2=0.991(图5B);MGF110-14L:y=0.152 7x+3.764 3,R2=0.993(图5C)。普通qPCR。p72:y=3.414x+1.461,R2=0.999(图5D)。综上所述,PMA对活病毒的扩增几乎无影响。
2.4 多重PMA-qPCR方法检测人工感染样品
利用多重PMA-qPCR方法与普通qPCR方法验证4份人工感染的ASFV阳性样品,结果如图6所示。当样品中全为ASFV活病毒时,多重PMA-qPCR结果与普通qPCR中ASFVp72基因的Ct值几乎一致,多重PMA-qPCR检测p72基因Ct值分别为26.1、26.75、26.3、26.75;普通qPCR检测p72基因Ct值分别为25.4、25.4、25.4、25.4。多重PMA-qPCR方法检测CD2v、基因Ct值分别为26.3、26.7、26.3、26.1;检测MGF110-14L基因的Ct值分别为26.3、26.5、26.3、26.3。统计学差异性分析显示,多重PMA-qPCR检测3个基因之间Ct值无显著差别(图6A,图6B)。当样品中均为ASFV灭活病毒时,普通qPCR反应仍能测得p72基因Ct值为28.0、28.0、28.0、28.0(图6C),而经PMA处理的多重PMA-qPCR方法测得p72、CD2v、MGF110-14L 3个基因Ct值,p72基因Ct值为36.5、36.8、36.4、36.7;CD2V基因Ct值为35.5、36.1、36.3、36.5;MGF110-14L基因Ct值为36.3、36.5、36.2、36.3(图6D),符合临床阴性样品标准检测线(Ct值≥35),说明灭活病毒的DNA被PMA抑制,不能继续扩增。显然,相比于普通qPCR方法本研究建立的多重PMA-qPCR方法更能有效区分样品中的ASFV活/灭活病毒。
2.5 多重PMA-qPCR方法检测实际样品
利用多重PMA-qPCR与基于p72基因建立的普通qPCR方法对23份阳性样本以及对应23份灭活样本进行检测。结果显示,普通qPCR方法对感染性活病毒样品与加热灭活的非感染性死病毒样品对p72基因的阳性检出率均为100%;本研究建立的多重PMA-qPCR方法只能在感染性活病毒样品检出p72基因,不能在加热灭活处理的非感染性死病毒样品检出p72基因。表明,本研究建立的多重PMA-qPCR方法能有效区分临床样品中的ASFV感染性与非感染性病毒(活/死病毒)。此外,对23份临床样品的多重PMA-qPCR检测发现,CD2V和MGF110-14L基因也均为100%阳性(图略),说明临床样品中未发现这两个基因的缺失株。
3 讨论
非洲猪瘟是由ASFV感染引起的猪的一种急性、热性、高度接触性传染病,具有跨国跨地区的传播能力,是养猪业的头号杀手[23]。中国是养猪大国,ASFV感染导致我国养猪业受到严重的影响[24],当务之急是建立完善的ASFV检测体系,做到高速、高效、精准地检测ASFV,使其病毒扼杀在传播初期。
目前,我国ASFV的检测仍以普通PCR和荧光定量PCR为主[25]。然而,这两种方法不能有效区分样本中的感染性与非感染性病毒[26]。PMA-qPCR技术在植物病原菌检测活菌的研究中被广泛应用[27-28],研究发现PMA能实现在不干扰活菌扩增的条件下实现对死菌扩增的抑制,这一现象弥补了普通PCR和荧光定量PCR的不足,很大程度上提高了病毒检测的准确性。
本研究建立的多重PMA-qPCR方法,当加入PMA浓度为10 μg/mL、暗孵育10 min、光照强度40 W、光照15 min时PMA能够抑制灭活病毒核酸扩增且对活病毒核酸扩增无影响,表明该方法能有效区分非洲猪瘟感染性活病毒与非感染性死病毒。通过对比普通qPCR与PMA-qPCR的灵敏度可知,两者的灵敏度均为102copies/μL;多重PMA-qPCR与普通qPCR方法验证人工感染样品,结果显示,当样品中全为ASFV活病毒时,多重PMA-qPCR结果与普通qPCR中ASFVp72基因的Ct差别不大;当样品中均为ASFV灭活病毒时,普通qPCR反应仍能测得p72基因,而经PMA处理的多重PMA-qPCR方法测得p72、CD2v、MGF110-14L基因Ct值,符合临床阴性样品标准检测线(Ct值≥35),说明灭活病毒的DNA被PMA抑制,不能继续扩增;将PMA-qPCR方法用于23份实际非洲猪瘟阳性样品的检测,结果显示,该方法检测临床样品中带有感染性活病毒的准确率为100%,说明多重PMA-qPCR方法能有效区分临床样品中具有感染性的非洲猪瘟活病毒。由于本研究建立的多重PMA-PCR方法还能检出CD2V和MGF110-14L基因,而这两个基因是ASFV的毒力基因,常用于构建ASFV疫苗缺失株的靶点;因此,多重PMA-qPCR方法有望用于野毒株与疫苗缺失株鉴别检测。
综上本研究建立的多重PMA-qPCR检测方法,具有特异性强和灵敏度高的特点,可高效检测ASFV活病毒,弥补了常规分子生物学检测方法的缺陷,解决了目前临床ASFV检测方法的不足,给养猪业带来更多的便捷,对ASFV早期诊断具有重大意义。
